



Contents
Scroll to:
 E. O. Mamedova,
E. G. Przhiyalkovskaya,
S. A. Buryakina,
E. V. Bondarenko,
A. M. Lapshina,
M. Yu. Pikunov,
Zh. E. Belaya,
G. A. Melnichenko
E. O. Mamedova,
E. G. Przhiyalkovskaya,
S. A. Buryakina,
E. V. Bondarenko,
A. M. Lapshina,
M. Yu. Pikunov,
Zh. E. Belaya,
G. A. Melnichenko https://doi.org/10.14341/probl13346
Scroll to:
Acromegaly is a neuroendocrine disorder caused by excessive production of growth hormone (GH). In the majority of cases the cause of acromegaly is a pituitary tumor producing GH. Cases of ectopic acromegaly are much rarer. Ectopic acromegaly occurs in cases of tumors which produce growth hormone-releasing hormone (GHRH) or extrapituitary tumors which produce GH. The main sources of excessive GHRH production are neuroendocrine tumors (NETs) of the lung or pancreas. Treatment of ectopic acromegaly consists of surgical removal of the source of GHRH hyperproduction and in cases where surgery is not an option, somatostatin analogues, pegvisomant, chemotherapy, immunotherapy or radiation therapy are used.
In this article three cases of ectopic acromegaly due to GHRH-producing lung NETs are presented, each of them being notable for a number of features. In the first two cases, clinical symptoms were mild, besides in the second case ectopic acromegaly was accompanied by primary hyperparathyroidism. In the third case ectopic acromegaly was accompanied by pituitary macroadenoma, and after surgical removal of the lung NET remission of acromegaly was not achieved. In all three cases, lung NETs were detected incidentally on radiologic chest screening for other conditions.
Mamedova E.O., Przhiyalkovskaya E.G., Buryakina S.A., Bondarenko E.V., Lapshina A.M., Pikunov M.Yu., Belaya Zh.E., Melnichenko G.A. Ectopic acromegaly due to bronchial neuroendocrine tumors: the first description in Russia of three clinical cases. Problems of Endocrinology. 2024;70(1):66-80. https://doi.org/10.14341/probl13346
Acromegaly is a severe chronic neuroendocrine disease resulting from excessive production of growth hormone (GH) [1]. More than 95% of cases of acromegaly are caused by GH-producing pituitary adenomas (both sporadic and familial forms) [2][3]. Сases of ectopic acromegaly are much rarer (less than 5%) [4]. The term “ectopic acromegaly” in fact encompasses three definitions. First, these are tumours that produce growth hormone-releasing hormone (somatoliberin, GHRH), which in turn stimulates excessive GH production by the intact pituitary gland. These include both hypothalamic tumours (hamartomas, gliomas, or gangliocytomas), which may lead to the development of hyperplasia and then GH-producing pituitary adenoma, and neuroendocrine tumours (NETs) or, rarely, GHRH-producing tumours of non-neuroendocrine nature of various localisations [5]. Second, there are GH-producing ectopic pituitary adenomas located outside the sella turcica – in the sphenoid sinus, petrous part of temporal bone, or nasopharynx [6]. Third, there are extracranial GH-producing tumours (e.g., pancreatic NETs [7], or non-Hodgkin lymphoma [8]). The first description of ectopic acromegaly dates back to 1949, when Goldman described a patient with a combination of pulmonary NET, pituitary tumour and bilateral adrenal lesions, suggesting that the tumour produced a factor that stimulated the growth of tumours in other organs [cited in 5]. The role of pulmonary tumour as a cause of acromegaly was first demonstrated by Altmann and Schütz, who in 1959 reported a patient with clinical manifestations of acromegaly who did not achieve remission after pituitary irradiation but improved significantly after resection of the pulmonary tumour [cited in 9]. In 1982, two independent groups isolated a protein that stimulates GH production (GHRH) from pancreatic tumour tissue in two patients with acromegaly [10][11], establishing a causal relationship between the development of acromegaly and a tumour of extra-pituitary localisation.
By 2012, Garby et al. published a literature review summarising data on 53 known cases of ectopic acromegaly [12]; Ghazy et al. published data on 98 cases [13]. In 2022, Zendran et al. published a review summarising data on 127 known cases [14]. Overall, 2/3 of cases of ectopic acromegaly are diagnosed in women [13], and the main sources of GHRH secretion in ectopic acromegaly are bronchial NETs (typical and atypical bronchial carcinoids) (up to 43%) and pancreatic NETs (up to 35%) [14]. Pheochromocytomas, paragangliomas, lymphomas, thymomas and some other tumours may occur in isolated cases [12–14]. Only in two cases GHRH-producing tumours were not of neuroendocrine nature (a diffuse large B-cell lymphoma [15] and adenocystic bronchial carcinoma [16]). Among intracranial sources of ectopic GHRH production, mixed gangliocytomas-adenomas of pituitary gland are more common [14].
The clinical picture of ectopic acromegaly does not differ from that of typical acromegaly, and it can be suspected in the absence of visualisation of pituitary adenoma on brain magnetic resonance imaging (MRI) or when diffuse pituitary hyperplasia is detected [17]. Identifying the source of GHRH production is the main challenge in diagnosing ectopic acromegaly, and the removal of the primary tumour is the main treatment option [13][14]. If radical removal of the tumour is impossible, long-acting somatostatin analogues, pegvisomant, chemotherapy, immunotherapy, and radiation therapy may be prescribed [13][14].
This article describes three clinical cases of ectopic acromegaly due to GHRH-producing bronchial NETs, each of which is remarkable for a number of features. In Russia, clinical cases of ectopic acromegaly have not been previously described.
A female patient S.N.Y., born in 1962, was first admitted to Endocrinology Research Centre at the age of 58. Approximately 6–7 years before she noticed for the first time some changes in her appearance (facial swelling, feet enlargement), weight loss, general fatigue, irritability. During a routine check-up by a gynaecologist in 2014, she was suspected of having acromegaly and a brain MRI was recommended. The brain MRI revealed a pituitary microadenoma. A hormone blood test revealed hyperprolactinaemia (baseline data were not provided), and therapy with cabergoline 0.25 mg once a week was prescribed. The therapy resulted in the normalisation of prolactin levels. In 2015, elevated concentrations of insulin-like growth factor 1 (IGF-1) and GH were detected for the first time, and acromegaly was diagnosed. At the follow-up MRI examination in 2016, a 5×5 mm pituitary microadenoma remained. In September 2016, due to sustained normoprolactinaemia, cabergoline was cancelled, and acromegaly therapy with somatostatin analogues was started: 20 mg long-acting octreotide once every 28 days intramuscularly, which she received for 6 months. During the treatment, the patient noticed an improvement of her condition, but IGF-1 remained elevated. Due to poor tolerance of octreotide (lump and hyperaemia at injection sites), the therapy was cancelled. In December 2016, hyperprolactinaemia up to 1,098 mU/L was detected again, and cabergoline therapy was periodically resumed and cancelled. No changes in the size of the pituitary adenoma during treatment with somatostatin analogues and cabergoline were noticed.
In February 2017, during a routine chest X-ray, a tumour lesion in the left lung was detected for the first time. Multislice computed tomography (MSCT) revealed a lesion in the hilum of the left lung with clear distinct contours 44×42×50 mm in size, of heterogeneous density with small calcium inclusions of 32–30 Hounsfield Units. The lesion was suspected to be malignant, and in May 2017 the patient’s local oncologist’s practice performed a typical left lung S4-5 segmentectomy and lymphadenectomy. Immunohistochemical (IHC) examination of the removed tumour found a highly differentiated Grade 1 bronchial NET with Ki-67 proliferation index under 2%, which is characteristic of a typical carcinoid. In the postoperative period, the patient noted a significant improvement of her condition: return of her former weight (weight gain of 18 kg), disappearance of facial swelling, reduction of general fatigue. Follow-up control established normalisation of IGF-1 and GH levels. MSCT found no evidence of tumour relapse. The patient still had hyperprolactinaemia: monomeric prolactin level was 1,897 mU/L, and a 4×5×5 mm pituitary microadenoma remained; the patient was periodically treated with cabergoline, with positive effect, and continued to be observed by an endocrinologist as she was diagnosed with acromegaly and no connection was established between the removal of the bronchial tumour and the achievement of acromegaly remission.
In 2020, the patient with acromegaly, pituitary microadenoma, bronchial NET, chromogranin A elevated to 169.12 μg/L (0–100) was suspected to have multiple endocrine neoplasia type 1 syndrome (MEN1) and was admitted to Endocrinology Research Centre. During in-patient examination, remission of acromegaly was confirmed: IGF-1 was 177.2 ng/ml (17–238); growth hormone nadir during oral glucose tolerance test (OGTT) was 0.274 ng/ml. On physical examination, mild manifestations of acromegaly were observed (Figure 1). Brain MRI revealed a lesion that was slightly hyperintense on T2 and hypointense on T1 WI, definitely not absorbing contrast agent, 14.4 mm wide, up to 7.3 mm high, and 2 mm in anteroposterior measurement. It was located at the border between anterior pituitary and posterior pituitary. The lesion was most consistent with Rathke’s cleft cyst. In the anterior right part of pituitary along the edge of the bottom and the right-side cavernous sinus, an area of slightly reduced content of contrast agent was found (5×5.7 mm, a microadenoma). The pituitary stalk was not displaced. A diffusely reduced MR signal was observed on T2 WI (Figure 2). An additional IHC study detected no GH expression in the removed bronchial carcinoid; diffuse GHRH expression was detected. (Figure 3 A, B).
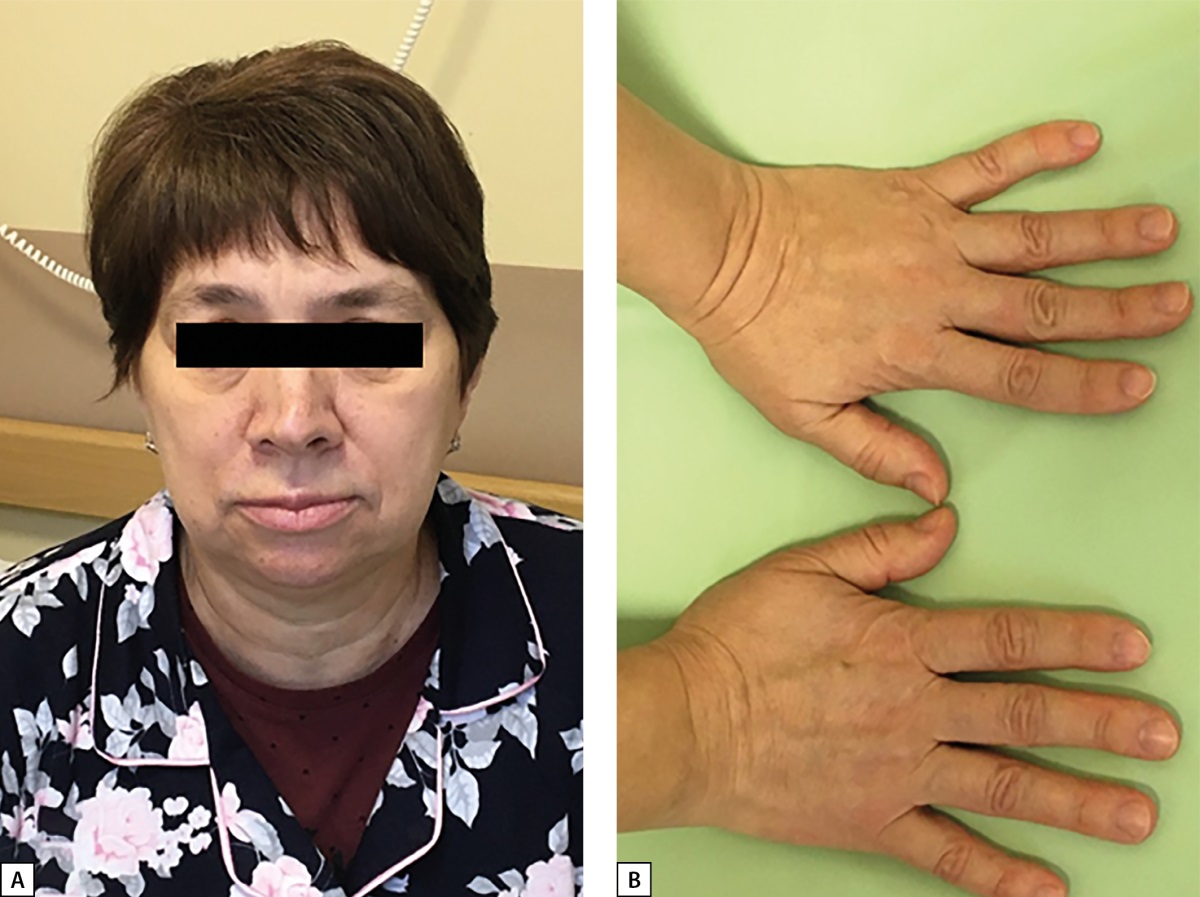
Figure 1: Patient S.N.Y. Typical changes in appearance in mild acromegaly at advanced age.
Slight enlargement of facial features (A); enlargement of hands (B).
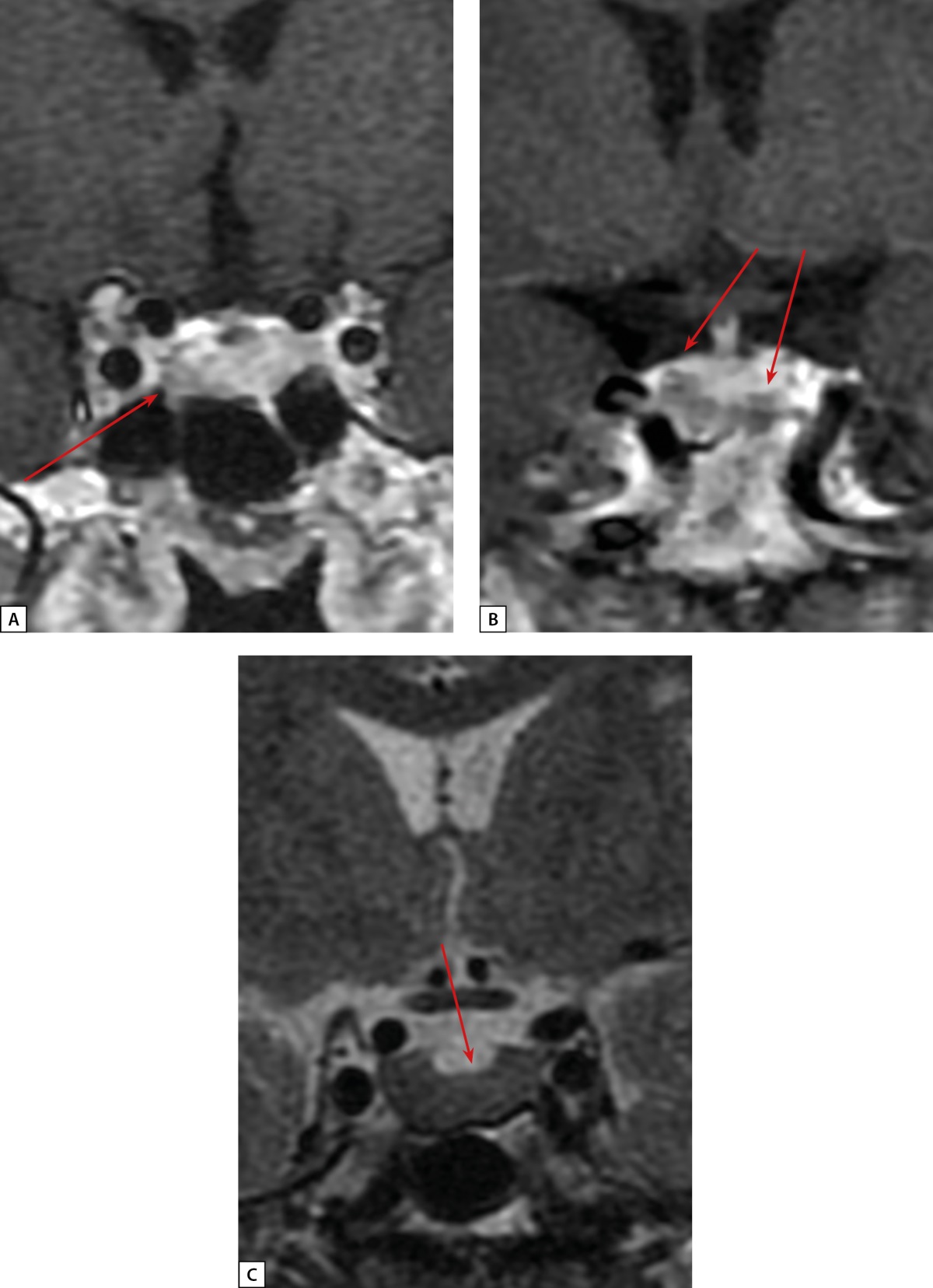
Figure 2: Patient S.N.Y. anterior pituitary cor MRI.
A: T1 WI with contrast agent. A microadenoma (see arrow) in the left anterior pituitary; B: T1 WI with contrast agent. Rathke’s cleft cyst (see arrows); C: T2 WI. A diffusely reduced MR signal on T2 WI from anterior pituitary tissue (see arrow)
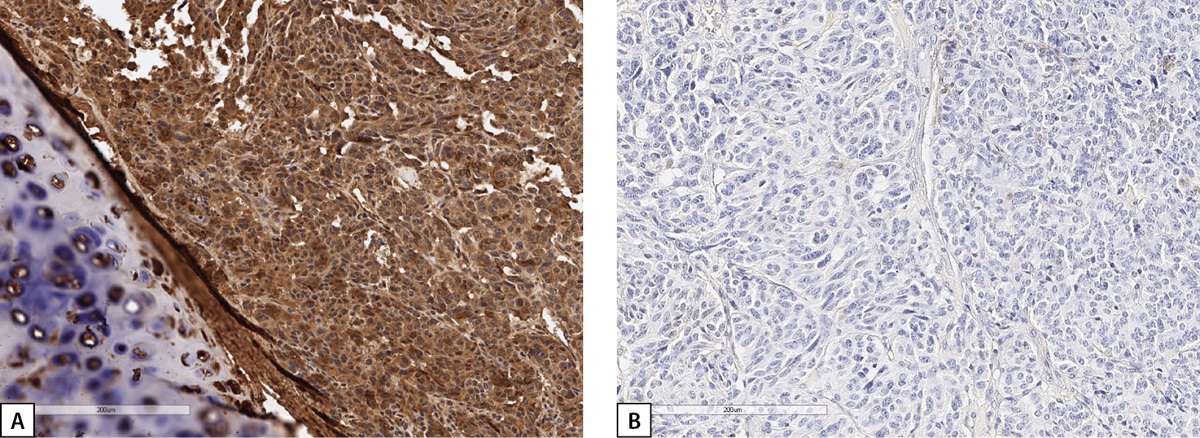
Figure 3: Immunohistochemical examination of Patient S.N.Y.’s bronchial carcinoid.
A: diffuse GHRH expression; B: no GH expression. Histological scans.
During examination for complications of acromegaly, there was no evidence of carbohydrate metabolism disorders (fasting blood glucose 4.88 mmol/L, two-hour blood glucose 5.8 mmol/L). Given a history of multinodular goitre, an ultrasound examination of the thyroid gland was performed, which revealed an increase in volume up to 27.7 ml with multiple nodular lesions with a maximum size up to 3.8 cm in diameter in the right lobe and up to 1.3 cm in the left lobe of the thyroid gland. A fine-needle aspiration biopsy confirmed a proliferative colloid goitre of varying degree (Bethesda II). Hormonal examination showed euthyroidism (TSH – 0.565 mIU/L; free T4 – 14.1 pmol/L (9.0–19.0); free T3 – 3.5 pmol/L (2.6–5.7), calcitonin – 3.41 pg/ml (0–4.8). No cardiovascular complications were found. Colonoscopy excluded any colon neoplasms. Due to complaints of back pain, a spinal radiography was performed, and a compression fracture of the Th12 vertebra was detected. The patient was diagnosed with osteoporosis of mixed origin, and antiresorptive therapy was prescribed.
After cancelling cabergoline and achieving acromegaly remission, persistent elevated monomeric prolactin up to 1,116 mIU/L (64–395) was noticed. To exclude possible secondary causes of prolactin level increase, the patient was examined for gynaecological diseases and no pathology was found. The patient’s fibrocystic breast changes could be a possible cause of hyperprolactinaemia. Given a moderate increase in the prolactin level, achievement of normoprolactinaemia after administration of small doses of cabergoline, and no growth of the pituitary adenoma in size, this pituitary microadenoma was considered non-functioning. Given the combination of pituitary microadenoma and a bronchial NET, screening examination for possible components of MEN1 syndrome was carried out: no data for primary hyperparathyroidism (PHPT), pancreatic or adrenal tumours were obtained. Blood test for chromogranin A – 2.1 nmol/L (under 2).
In 2022, the patient continued to be monitored in Endocrinology Research Centre. Persistent acromegaly remission was maintained, and positive changes in bone mineral density were observed after administration of zoledronic acid.
A female patient D.I.E., born in 1959, was first admitted to Endocrinology Research Centre at the age of 60. Her complaints included marked general weakness, headaches, joint pain, sweating, weight loss, and facial swelling. The patient considered herself to have been unwell for the last two years, ever since an examination after the flu revealed an increase in blood glucose up to 9.3 mmol/L. Later, glycaemic parameters normalised and diet therapy was recommended. Due to general malaise, the patient visited a local endocrinologist’s practice, and the examination revealed an increase in IGF-1 level up to 349 ng/ml (normal up to 200). Brain MRI showed marked irregularity of the anterior pituitary structure on the left side; thus, a pituitary adenoma could not be excluded. In the middle part of the pituitary, a T2 WI hyperintense structure was observed (3.8×1.5 mm), which did not absorb contrast agent (Rathke’s cleft cyst). The MR signal from the anterior pituitary was slightly reduced on T2 WI (Figure 4 a, b).
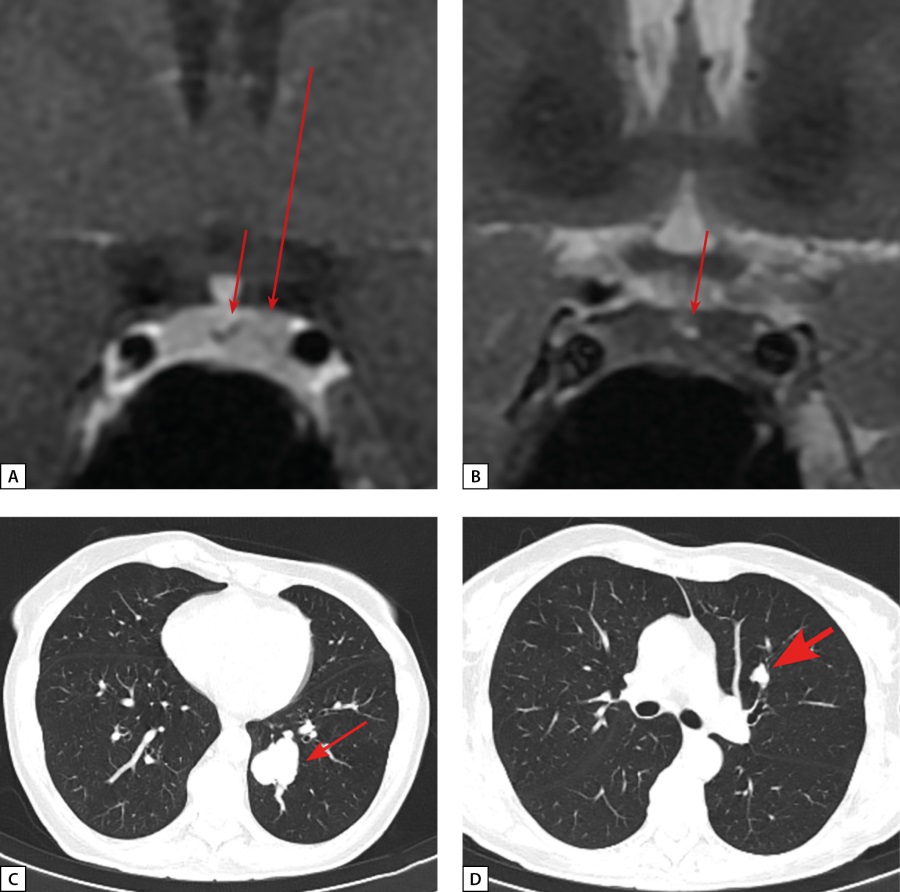
Figure 4: Patient D.I.E. MRI and CT scans.
A: anterior pituitary cor MRI, T1 WI with contrast agent. Rathke’s cleft cyst (see arrow). An area of slightly reduced contrast agent absorption in the left side of anterior pituitary (see long arrow); B: Rathke’s cleft cyst (see arrow). Diffusely reduced MR signal on T2 WI from anterior pituitary tissue; C: lung MSCT; pulmonary mode, native phase, axial projection. A neuroendocrine tumour in S10 of the left lung (see short arrow); D: lung MSCT; pulmonary mode, native phase, axial projection. A lesion in S3 of the left lung with intra- and peribronchial spreading – a seeding site (bold arrow).
Upon admission to Endocrinology Research Centre, active acromegaly was confirmed: IGF-1 at 591.1 ng/ml (15–230); GH nadir of 2.53 ng/ml during OGTT. Notably, clinical signs of acromegaly were subtle: a slight facial oedema and the enlargement of the nose and fingers. Also during the examination the PHPT diagnosis was verified for the first time: parathyroid hormone was elevated to 78.8 pg/ml (15–65), total calcium was at 2.51 mmol/L (2.15–2.55); ultrasound examination of parathyroid glands revealed a 1.2×0.5×0.7 cm lesion in the right inferior parathyroid gland. PHPT complications included osteopenia in 33% of the radial bone to -2.0 SD by T-score. A transnasal transsphenoidal adenomectomy was performed in February 2019. GH nadir during OGTT in the early postoperative period was 0.89 ng/ml. The removed material included anterior pituitary tissue with extensive areas of oxyphilic cells, and the presence of a pituitary tumour was suspected. However, additional slices with silver impregnation revealed a preserved network of reticulin fibres with microfoci of dilated acinuses, which corresponds to anterior pituitary hyperplasia. On immunohistochemical examination, most of the cells were GH-immunopositive (Figure 5).
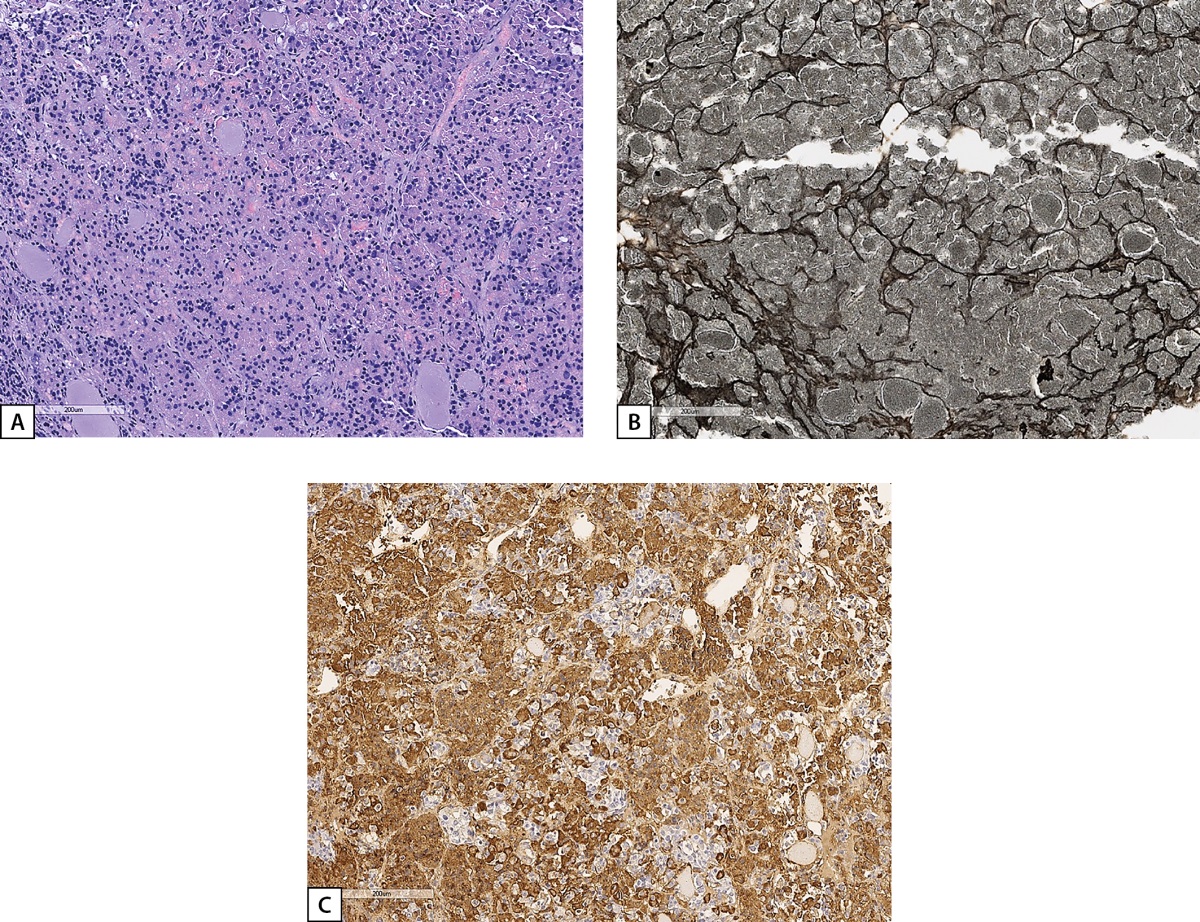
Figure 5: Histological and IHC study of the material surgically removed from Patient D.I.E.’s pituitary.
A: anterior pituitary tissue with extensive areas of oxyphilic cells (haematoxylin and eosin); B: preserved network of reticulin fibres with microfoci of dilated acinuses (silver impregnation); C: diffuse GH staining of anterior pituitary cells (an HIS reaction with GH antibodies). Histological scans.
Given the combination of acromegaly and PHPT, in order to exclude MEN1 syndrome, we performed high-throughput parallel sequencing of a panel of candidate genes (AIP, CASR, CDKN1A, CDKN1B, CDKN1C, CDKN2A, CDKN2C, CDKN2D, DICER1, GNAS, CDC73, MEN1, POU1F1, PRKAR1A, PRKCA, PTTG2, SDHA, SDHB, SDHC, and SDHD (total coverage of coding exons: 96.7%)), and the results showed no mutations.
Three months after surgical intervention, IGF-1 remained elevated at 389.2 ng/ml (89–255). When the patient was readmitted to Endocrinology Research Centre one year after the surgical intervention, the absence of acromegaly remission was confirmed: IGF-1 was 414.5 ng/ml (17–238). However, contrast brain MRI did not provide conclusive evidence of residual pituitary adenoma tissue. Long-acting octreotide therapy was recommended, which resulted in normalisation of IGF-1 level to 196.6 ng/ml (93–224).
At the age of 61, the patient had COVID, and chest MSCT revealed an irregularly shaped 32×32 mm lesion in S10 of the left lung. It had clear, polycyclic contours and density of 34 Hounsfield Units (Figure 4 c, d). The lesion was located in the lumen of the posterior basal bronchus, as well as peribronchially in the parenchyma of S10. Partial consolidation of the parenchyma of the segment distal to the lesion (subsegmental atelectasis) was observed. In S3 of the left lung, there was a second lesion with clear irregular contours, 16×13 mm in size and density of 56 Hounsfield Units. A lesion was also located in the lumen of the anterior bronchus and in the parenchyma of S3. Regional lymph nodes were not enlarged. A conclusion was made: the lesion in S10 of the left lung was most consistent with NET; subsegmental atelectasis in S10 of the left lung was distal to the lesion; the lesion in S3 was a seeding site. A videothoracoscopic inferior lobectomy on the left side and a lymphodissection on the left side were performed at the patient’s local clinic. Histological examination revealed atypical carcinoid of Grade 2 with maximum tumour size 30 mm. No tumour growth was found in the bronchus resection margin, vessel resection margins, visceral pleura, and in any of the examined specimens.
Due to suspected ectopic acromegaly, histological specimens were re-examined at Endocrinology Research Centre and an IHC study of the removed tumour was performed. A conclusion was made: in the presented histological specimens stained with haematoxylin and eosin, a tumour was found with evidence of neuroendocrine differentiation with two mitoses, evidence of invasive growth into the bronchi, seven lymph nodes with reactive changes, without elements of tumour growth in the pulmonary tissue. In the presented histological specimens stained with antibodies to Ki-67, TTF-1, cytokeratin AE1/3 and CD56, expression of TTF-1, cytokeratin AE1/3, CD56 was found in the tumour cells, Ki-67 label index was 13.2%. An IHC examination with antibodies to GHRH revealed a focal GHRH expression of moderate to high intensity in the majority of tumour cells; reaction to GH was negative (Figure 6).
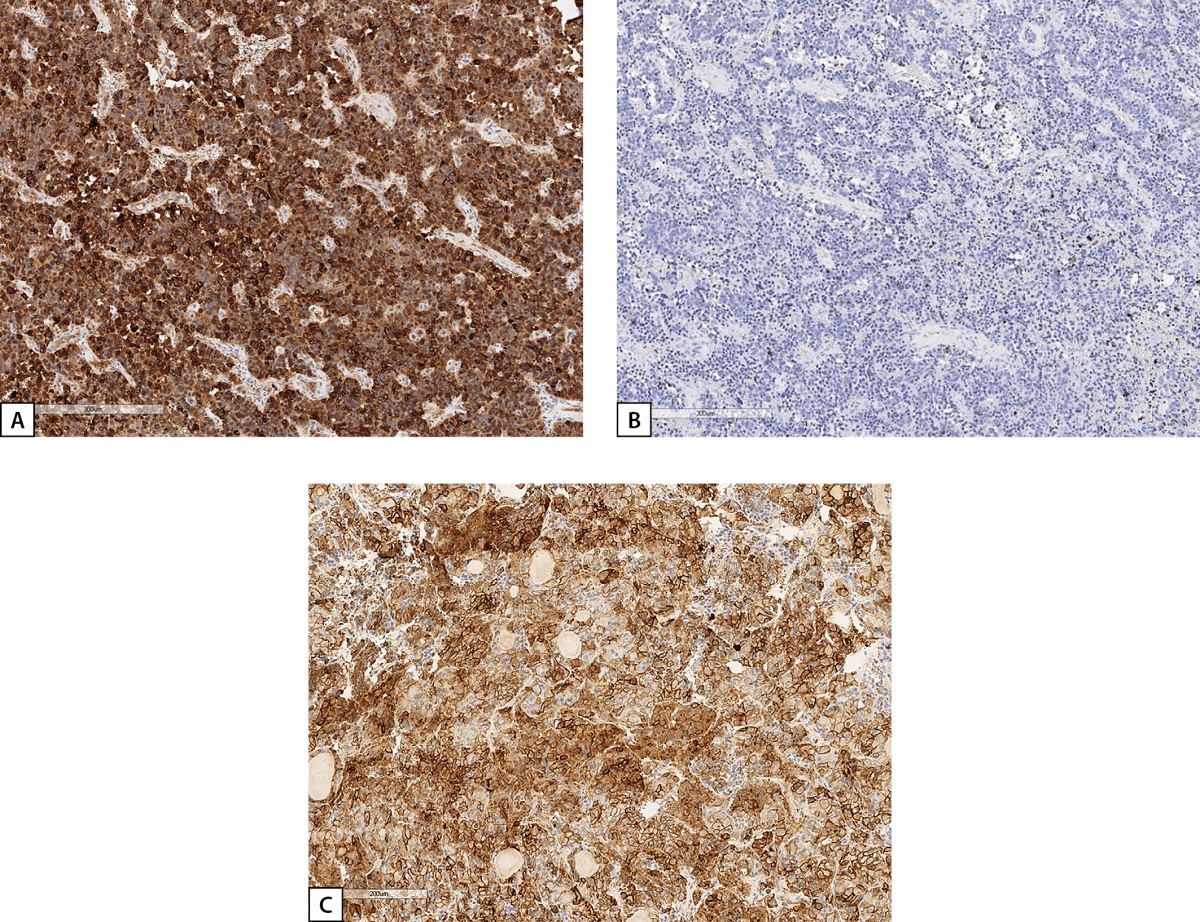
Figure 6: IHC study of Patient D.I.E.’s bronchial carcinoid.
A: diffuse GHRH expression; B: no GH expression; C: positive expression of 2A somatostatin receptors (SSTR 2A) (3 points). Histological scans.
Thus, the diagnosis of ectopic acromegaly was established based on the results of histological and IHC examination. A decision was made to temporally discontinue somatostatin analogues; following the discontinuation, IGF-1 remained within the reference values (136 ng/ml (17–238)). Positron emission CT with 68Ga-DOTATATE/DOTANOC performed 9 months after the surgical intervention revealed no signs of tumour growth. Two and a half years after the surgical intervention, the patient remains in acromegaly remission. During the follow-up, parathyroid hormone was still elevated up to 102.5 pg/ml (15–65), with total calcium at 2.52 mmol/l (2.15–2.55) and ionised calcium at 1.33 mmol/l (1.03–1.29); no secondary causes of parathyroid hormone elevation were found. Given the mild course of PHPT (absence of osteoporosis, urolithiasis, no decrease in the glomerular filtration rate, normocalciuria), regular follow-up examinations were chosen.
A female patient S.N.A., born in 1978, was first admitted to Endocrinology Research Centre at the age of 41 with complaints of headache, back pain, joint pain, facial swelling, hair loss, weight gain, menstrual cycle irregularities (delays). From the age of 37 she began to notice enlargement of the feet, hands and nose, fingertip numbness, and menstrual cycle delays. At the age of 40, she was seen by an endocrinologist at her local medical practice, and acromegaly was suspected during examination. GH level was 8.29 ng/ml (0.5–7), but no IGF-1 data were available at that time. Contrast brain MRI revealed a 25×30×33 mm pituitary macroadenoma with endo-, supra-, infra-, and latero- (into the left cavernous sinus, Knosp Grade 3) sellar growth. It compressed the optic nerve chiasm (Fig. 7a). Iron deficiency anaemia due to heavy menstruation associated with uterine myoma was also detected at that time. At the age of 41 the patient underwent transnasal transsphenoidal adenomectomy at her local clinic. Histological examination found a “large-cell pituitary adenoma”. After the surgery, secondary hypothyroidism developed, but acromegaly remission was not achieved, thus the patient was referred to Endocrinology Research Centre. Before admission, due to complaints of persistent cough, the patient underwent a chest MSCT, which revealed a 15×12 mm lesion in the upper lobe of the left lung, which the oncologist considered benign on the basis of CT phenotype.
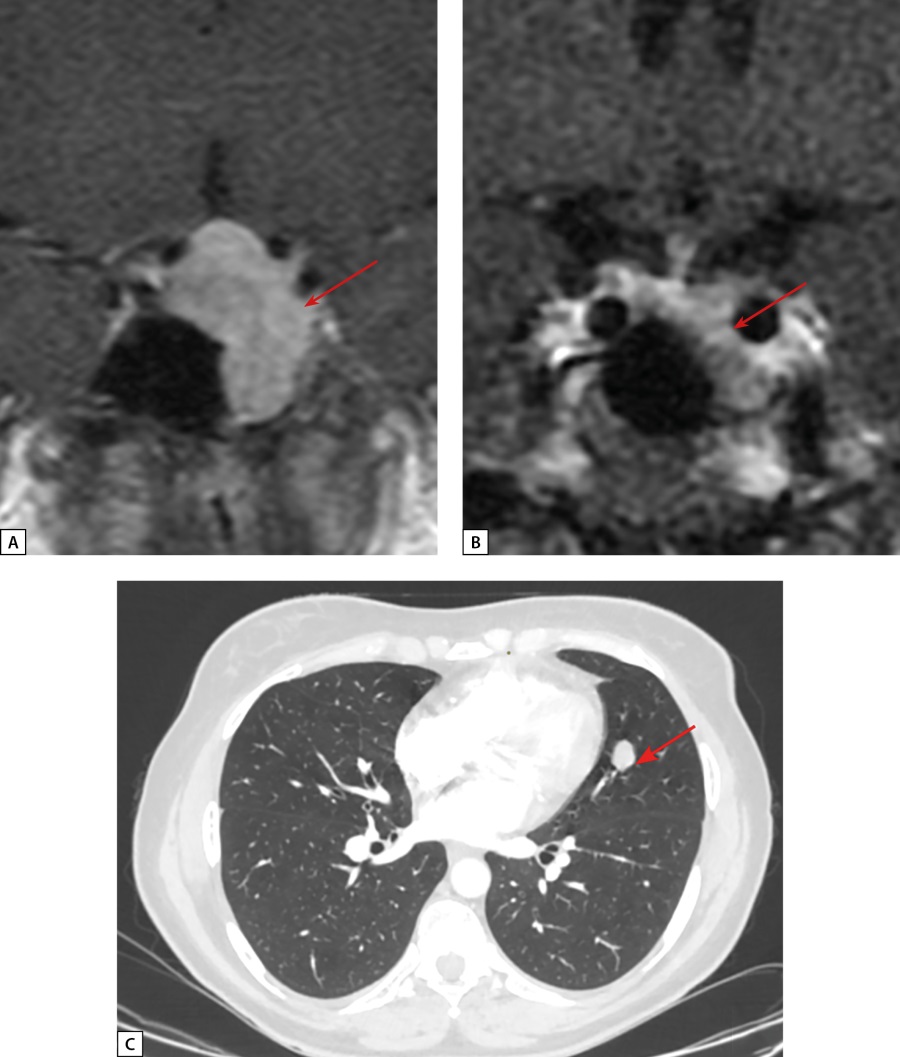
Figure 7: Patient S.N.A. MRI and CT scans.
A: pre-surgery pituitary MRI coronal T1 WI contrast. A pituitary macroadenoma (see arrow); B: post-surgery pituitary MRI coronal T1 WI contrast. A pituitary macroadenoma (see arrow), decreased in size; C: lung MSCT; pulmonary mode, arterial phase, axial projection. A neuroendocrine tumour in S4 of the left lung (see arrow).
Upon admission to Endocrinology Research Centre the patient’s examination confirmed active acromegaly: IGF-1 at 538.9 ng/ml (64–395); there was no GH suppression during OGTT (GH nadir – 1.03 ng/ml). Contrast brain MRI revealed a cystic-solid lesion 25x25x30 mm in the sella turcica (left side), in the sphenoid sinuses, more on the left side, in the left cavernous sinus (Knosp Grade 3). There was uneven absorption of the contrast by the lesion. Compared to MRI images made prior to transnasal transsphenoidal adenomectomy, new images showed moderate reduction of vertical size (distance to the optic chiasm: 5 mm) and spreading to the lateral part of the sphenoid sinus. Conclusion: pituitary macroadenoma with endo-, infra-, and latero- (S) sellar growth. The patient was consulted by a neurosurgeon, and repeated transnasal transsphenoidal adenomectomy was performed. Morphological and IHC examination revealed densely granulated somatotroph adenoma (Figure 8). GH nadir of 0.6 ng/ml was achieved during OGTT in the early postoperative period. Subsequently, long-acting octreotide 20 mg once every 28 days intramuscularly was prescribed by the patient’s local physician due to the absence of GH suppression during OGTT. Repeated examination at Endocrinology Research Centre one year post-surgery, after a period of therapy with long-acting octreotide, revealed IGF-1 elevation up to 332.4 ng/ml (51–271). Contrast brain MRI found a cystic-solid lesion of heterogeneous structure with uneven contrast absorption located in the sella turcica, with infrasellar growth into the sphenoid sinus, laterosellar spread into the left cavernous sinus (Knosp Grade 3), antesellar spread to the ethmoidal labyrinth, with maximum size: vertical – 18 mm, transverse – 25 mm, anteroposterior – 28 mm. Suprasellar cistern was not narrowed, nor was the optic nerve chiasm compressed. Compared to the previous examination, there was a decrease in the tumour size (Figure 7b). Given the lack of effect after two surgical interventions, the patient was recommended to increase the dose of long-acting octreotide to 30 mg once every 28 days intramuscularly.
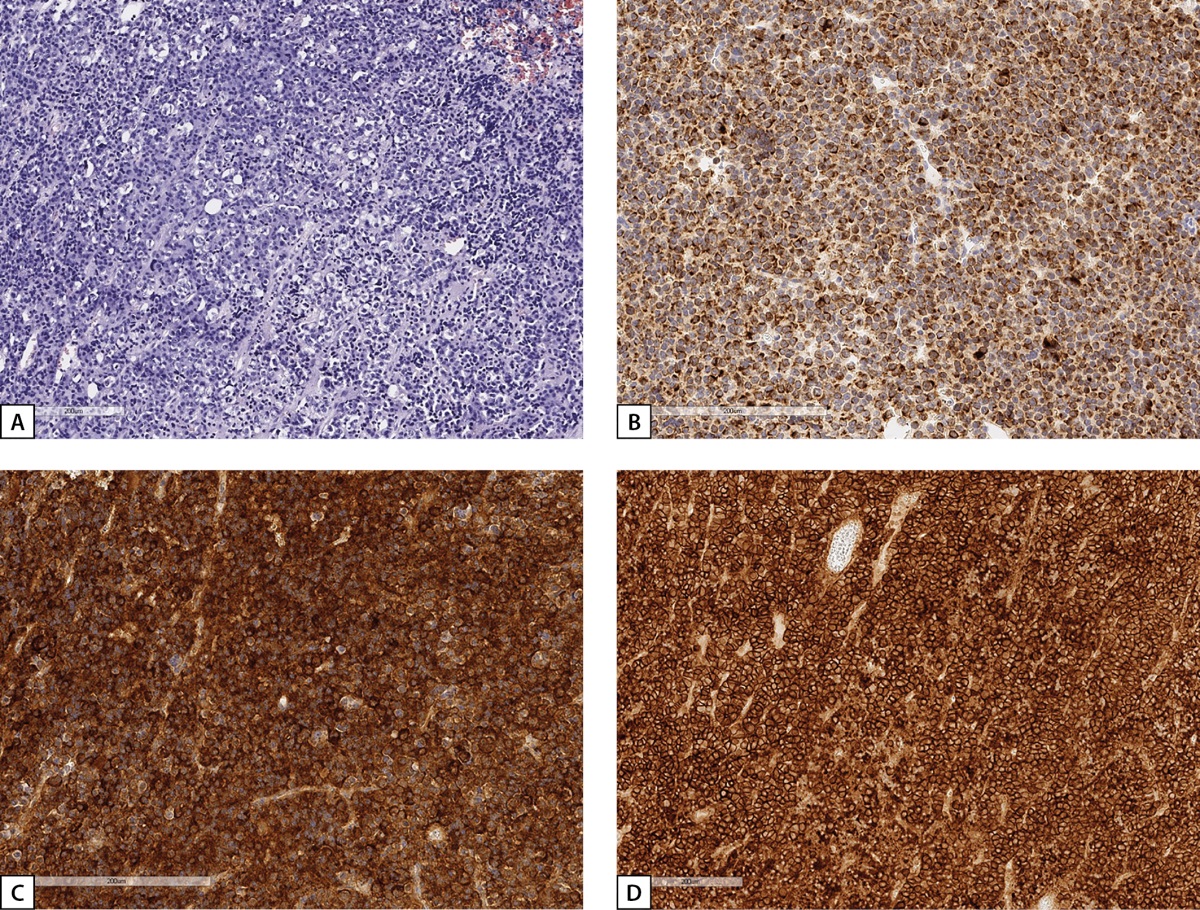
Figure 8: Histological and IHC study of the material surgically removed from Patient S.N.A.’s pituitary tumour.
A: a solid tumour of chromophobe cells (haematoxylin-eosin); B: irregular cytoplasmic staining with low-molecular-weight cytokeratin CAM 5.2 clone (IHC reaction); C: pronounced diffuse GH expression in tumour cells (IHC reaction); D: pronounced expression of 2A somatostatin receptors, IRS=12 (HIS reaction). Histological scans.
At the age of 44, the patient was readmitted to Endocrinology Research Centre for a follow-up examination. At the time of admission, she was receiving long-acting octreotide 40 mg once in 28 days (the dose was increased by her local physician due to the lack of IGF-1 level normalisation). During examination, the absence of acromegaly remission was confirmed: IGF1 was at 300.8 ng/ml (51–271). Contrast brain MRI found a cystic-solid lesion of heterogeneous structure in the sella turcica, in the sphenoid sinus (spreading to the posterior labyrinth). The lesion was protruding from anterior pituitary and unevenly absorbing contrast and had the same size: vertical – 18 mm, transverse – 21 mm, anteroposterior – 25 mm, without any changes vs. the previous examination. Given a lung lesion in the history, a contrast chest MSCT was performed: in the left S4 lung parenchyma a perivascular solid lesion 15×12×19 mm was seen with smooth and slightly bulging contours, moderately absorbing contrast agent (native/ arterial/ venous/ deferred phases: 13/ 15/ 69/ 79 Hounsfield Units). Conclusion: the lesion was most consistent with NET (Figure 7c). A S4–5 bisegmentectomy on the left side with groups 3, 5, 7, 9 lymphodissection was performed. Morphological examination detected a dense, homogeneous pale yellow 1.6×1.5×1.3 cm tumour nodule located within the lung lobe tissue. Microscopically, the tumour nodule was a dense cellular formation with scanty, slightly eosinophilic, vaguely contoured cell cytoplasm and small oval uniform nuclei. The cells formed solid-alveolar structures; no foci of necrosis were found. There were three mitoses in 50 fields of view at ×400 magnification. The tumour involved the bronchus wall, grew under the epithelium of its mucous membrane and narrowed the bronchus lumen. The nodule had a clear border with the surrounding pulmonary tissue, without a visible pseudocapsule. A IHC study revealed tumour cells to have diffuse expression of chromogranin A (clone DAK-A3, DAKO), Ki-67 (clone SP6, Cell Marque) – nuclear expression in less than 1% of cells. Conclusion: typical carcinoid of the left lung, Grade 1, with a lesion of the upper lobe subsegmental bronchus wall, without invasive elements in the resection margins and without metastases in seven regional lymph nodes.
Given the findings, ectopic acromegaly was suspected. IHC examination revealed diffuse GHRH expression, which confirmed ectopic acromegaly diagnosis (Figure 9).
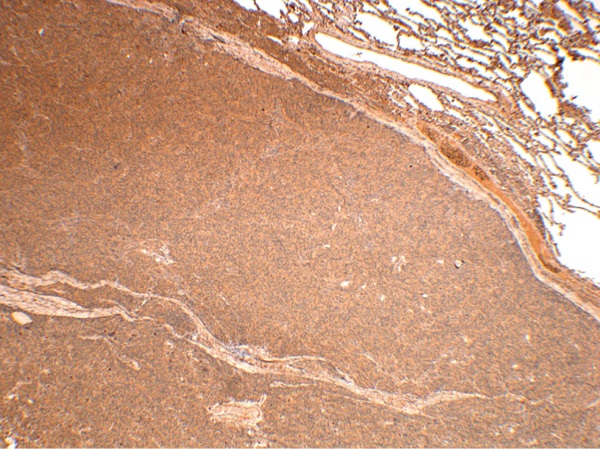
Figure 9: IHC study of Patient S.N.A.’s bronchial carcinoid. Diffuse GHRH expression. Magnification: 40×.
However, IGF-1 levels remained elevated in the postoperative period, thus necessitating continued therapy with somatostatin analogues. Table 1 compares the parameters of ectopic acromegaly cases described above.
Table 1: Parameters of patients with ectopic acromegaly
|
Case 1 |
Case 2 |
Case 3 |
|
|
Age at the time of diagnosis |
58 |
60 |
41 |
|
Sex |
Female |
Female |
Female |
|
IGF-1 |
N/A |
591.1 ng/ml (15–230) |
538.9 ng/ml (64–395) |
|
Basal GH |
N/A |
N/A |
8.29 ng/ml (0.5–7) |
|
GH nadir during OGTT (under 0.4 ng/ml) |
N/A |
2.53 ng/ml |
1.03 ng/ml |
|
Brain MRI data |
A lesion in anterior pituitary, consistent with anterior pituitary microadenoma and Rathke’s cleft cyst |
Pronounced irregularity of anterior pituitary structure; suspected anterior pituitary microadenoma; 3.8×1.5 mm Rathke’s cleft cyst |
A 25×30×33 mm pituitary macroadenoma with endo-, supra-, infra-, and latero- (into the left cavernous sinus, Knosp Grade 3) sellar growth; the lesion is compressing the optic nerve chiasma |
|
Source of ectopic acromegaly |
A lesion in the left pulmonary hilum; size: 44×42×50 mm; Histological test: a highly differentiated Grade 1 bronchial NET with Ki-67 proliferation index under 2%, which is characteristic of a typical carcinoid |
A 32×32 mm lesion in S10 of the left lung, with a density of 34 Hounsfield Units; a seeding site in S3 of the left lung, with clear irregular contours, 16×13 mm in size and density of 56 Hounsfield Units. Histological test: an atypical carcinoid, G2 |
A 15×12 mm lesion in the upper lobe of the left lung. Histological test: a typical carcinoid in the left lung; Grade 1; lesion of the upper lobe subsegmental bronchus wall, without invasion elements in the resection margins and without metastases in seven regional lymph nodes |
This article describes three clinical cases of ectopic acromegaly in women due to bronchial NETs, which is consistent with the literature, according to which they are the most frequent localisation of GHRH-producing NETs, and that the disease is more frequent in women [13][14]. Each case is notable for a number of features. Case 1 presented with weight loss, which is not typical of acromegaly but may be a symptom of NET. Case 2 was characterised by mild clinical manifestations of acromegaly, although, as mentioned above, clinical manifestations of classical and ectopic acromegaly are thought to be similar [17]. In all three patients, the diagnosis of ectopic acromegaly was made ex post facto: in Case 1, after retrospective analysis of the patient’s medical history, in Cases 2 and 3 – after failure to achieve acromegaly remission after transnasal adenomectomy and after incidental detection of lung lesions when screening for other conditions.
The question remains relevant: what data could provide a basis for an endocrinologist to suspect ectopic GHRH production in a patient with clinical signs of acromegaly? In 2022, Potorac et al. retrospectively analysed brain MRI data in 30 ectopic acromegaly patients in order to identify distinctive features of pituitary imaging in such cases [17]. The results showed pituitary hyperplasia in 24 patients, in two cases the vertical size of the pituitary gland was slightly increased relative to normal, and in four cases pituitary gland size was normal. Based on the analysis of MRI images, the authors conclude that pituitary in ectopic acromegaly patients is slightly to moderately enlarged in size; pituitary is hypointense on T2-weighted images, and normal pituitary tissue is not visualised on MRI. In addition, there is no invasion into cavernous sinuses or compression of the chiasm. The authors highlight the following factors as suggesting that ectopic acromegaly could be suspected: hypointense pituitary image on T2-weighted images (in 83.3% of cases), pituitary hyperplasia (80%), average pituitary height of about 9.5 mm (rarely more than 18 mm), normal pituitary tissue is not visualised, no invasion into the cavernous sinuses/sphenoid sinus, no pituitary stalk deviation, no changes in the sellar floor. In addition to MRI characteristics, the authors note that ectopic acromegaly is more frequent in females (73.3%), and in males the disease occurs at a younger age. In addition, in 50% of cases, acromegaly was diagnosed before a NET; the most frequent localisations are lungs (56.7%) and pancreas (33.3%) [17].
Thus, the absence of pituitary adenoma MRI visualisation in a patient with acromegaly allows us to suspect ectopic acromegaly. Nevertheless, a number of studies indicate that this is not always the case. In a study by Lonser et al. among 190 acromegaly patients, there were six (three males and three females) in whom no pituitary adenoma was visualised on brain MRI. Three patients underwent MRI using the VIBE sequence (volumetric interpolated breath-hold examination), which revealed a 4 mm pituitary adenoma in one patient. All patients underwent endoscopic transnasal revision of the pituitary gland, and microadenomas were detected in all of them. Morphological and IHC studies confirmed somatotroph adenoma in all cases, and all patients achieved biochemical acromegaly remission after surgery [18]. Similar observations have been reported by others [19].
In the described cases, the first patient had a pituitary microadenoma (most likely non-functioning); the second patient had pituitary hyperplasia, which was initially considered a pituitary microadenoma. Only Case 3 had a pituitary macroadenoma, which is not typical for ectopic acromegaly. It is possible that the pituitary macroadenoma developed due to prolonged exposure of the pituitary gland to GHRH, and this prevented acromegaly remission after surgical removal of the bronchial NET. We found no cases of a combination of ectopic acromegaly with GH-producing pituitary macroadenoma in one patient in the literature.
Measuring plasma GHRH levels may have diagnostic significance in suspected ectopic acromegaly. Various researchers have suggested the following plasma GHRH levels at which it may be suspected: over 300 ng/L [20] or over 250 ng/L [12]. In general, there are commercial kits for GHRH measurement; the most common one is ELISA [14]. However, given the rarity of the disease and the difficulty in obtaining such kits, it seems reasonable to perform diagnostic imaging (chest and abdomen) if pituitary adenoma is not visible on brain MRI, both initially and in failure to achieve postoperative acromegaly remission and also in cases where residual tumour tissue is not visible on brain MRI after surgery.
In two of the three cases described (Cases 1 and 2), surgical treatment (removal of bronchial NETs) resulted in acromegaly remission. In general, surgical intervention is the first-line treatment for ectopic acromegaly, as it allows complete removal of the source of GHRH hyperproduction. If radical removal of the tumour is not possible, various other treatments are used. Long-acting somatostatin analogues can be used due to the presence of somatostatin receptor type 2 expression in NETs [21]. If there is no expression of these receptors in tumour tissue, somatostatin analogues may still have a partial effect due to their impact on pituitary somatotrophs, which reduces GH expression. In some cases, radiation therapy, as well as chemotherapy, immunotherapy, embolisation of metastases, and administration of pegvisomant have been used [14].
The combination of ectopic acromegaly and PHPT in Case 2 is noteworthy. Based on the combination of acromegaly and PHPT, the patient was clinically suspected to have MEN1 syndrome. A review by Zendran et al. indicated that 23 patients with ectopic acromegaly and MEN1 syndrome have been described in the literature, with genetic analysis confirming the diagnosis in 19 of them [14]. Notably, 18 cases of NET were localised in the pancreas, and one case in the thymus, but none in the lungs. In addition to pancreatic NETs, most patients had PHPT [14], and some patients also had pituitary adenomas (gonadotroph adenoma [20], GH/prolactin-secreting pituitary adenoma [12], null-cell adenoma [23]). Thus, in MEN1 patients, diagnosis of ectopic acromegaly due to GHRH-producing pancreatic NET may be difficult due to a possible combination with pituitary adenoma (non-functioning or otherwise) within this syndrome. In general, a combination of pituitary adenoma and PHPT in a patient allows clinical diagnosis of MEN1 syndrome [24]. If no mutation in MEN1 gene is detected, this case can be considered as a phenocopy of MEN1 syndrome, and very rarely mutations in CDKN1B gene (multiple endocrine neoplasia syndrome type 4, MEN4) can be detected in such patients [25]. In our patient (Case 2), no mutations in MEN1 or CDKN1B were detected. Given no aggravated family history of acromegaly and the fact that acromegaly is associated with increased incidence of some neoplasms [27], it is possible to assume either that the parathyroid adenoma developed due to excessive IGF-1 exposure, or a coincidental combination of several endocrine tumours in the same patient.
As of 1 May 2023, Russia’s National Hypothalamic-Pituitary Tumour Registry had records on 5,726 patients with acromegaly. International data suggests that 5% of acromegaly patients will have an extra-pituitary cause of the disease [4], and thus over 250 residents of Russia are likely to have acromegaly caused by ectopic GH or GHRH secretion. However, no such cases have been reported by Russian authors in the literature. The reasons of untimely (retrospective) diagnosis of ectopic acromegaly may include low awareness of doctors, including endocrinologists, lack of specific clinical manifestations, inaccessibility of laboratory GHRH measurement, high sensitivity of NETs to therapy with somatostatin analogues, lack of practicians’ vigilance with regard to patients with acromegaly who have not achieved remission after neurosurgical treatment involving radical removal of pituitary adenoma.
Acromegaly is relatively rare in the general population, and ectopic acromegaly occurs in only about 5% of cases. Nevertheless, endocrinologists need to suspect ectopic acromegaly if pituitary adenoma is not visible on brain MRI or when detecting a diffuse pituitary hyperplasia that is hypointense on T2 WI. Diagnostic search should be started with the exclusion of bronchial and pancreatic NETs, and in case of pancreatic NETs, MEN1 syndrome should be suspected. Surgical treatment is the first-line method as it potentially enables a complete removal of the source of GHRH hyperproduction and leads to acromegaly remission. In the absence of ectopic acromegaly remission after surgical treatment, it is advisable to prescribe various options of drug treatment.
Source of funding. Data collection and analysis and manuscript preparation were funded by the Russian Science Foundation (Project 19-15-00398).
Conflict of interest. The authors hereby declare they have had no apparent or potential conflict of interest related to the content of this publication.
Authors’ contribution. Every author has approved the final version of the text prior to publication and agreed to accept responsibility for all aspects of this study, which implies due investigation and resolution of any issue related to the accuracy or integrity of any part thereof.
Patients’ consent. The patients have voluntarily given their informed consent for the publication of their personal medical information in anonymised form in Problems of Endocrinology.
1. Belaya ZE, Golounina OO, Rozhinskaya LY, et al. Epidemiology, clinical manifestations and efficiency of different methods of treatment of acromegaly according to the United Russian Registry of Patients with Pituitary Tumors. Problems of Endocrinology. 2020;66(1):93-103. (In Russ.). doi: https://doi.org/10.14341/probl10333
2. Sakhnova EE, Przhiyalkovskaya EG, Belaya ZE, Melnichenko GA. Discordant parameters of insulin-like growth factor 1 and growth hormone in the diagnosis and monitoring of acromegaly. Problems of Endocrinology. 2022; 68(1):40-48. (In Russ.). doi: https://doi.org/10.14341/probl12791
3. Mamedova E, Vasilyev E, Petrov V, et al. Familial acromegaly and bilateral asynchronous pheochromocytomas in a female patient with a MAX mutation: a case report. Front Endocrinol (Lausanne). 12:683492. doi: https://doi.org/10.3389/fendo.2021.683492
4. Chanson P, Salenave S. Acromegaly. Orphanet J Rare Dis. 2008; 3:17. doi: https://doi.org/10.1186/1750-1172-3-17
5. Sano T, Asa SL, Kovacs K. Growth hormone-releasing hormone-producing tumors: clinical, biochemical, and morphological manifestations. Endocr Rev. 1988; 9(3):357-373. doi: https://doi.org/10.1210/edrv-9-3-357
6. Ramírez C, Hernández-Ramirez LC, Espinosa-de-los-Monteros AL, et al. Ectopic acromegaly due to a GH-secreting pituitary adenoma in the sphenoid sinus: a case report and review of the literature. BMC Res Notes. 2013; 6:411. Published 2013 Oct 12. doi: https://doi.org/10.1186/1756-0500-6-411
7. Melmed S, Ezrin C, Kovacs K, et al. Acromegaly due to secretion of growth hormone by an ectopic pancreatic islet-cell tumor. N Engl J Med. 1985; 312(1):9-17. doi: https://doi.org/10.1056/NEJM198501033120103
8. Beuschlein F, Strasburger CJ, Siegerstetter V, et al. Acromegaly caused by secretion of growth hormone by a non-Hodgkin’s lymphoma. N Engl J Med. 2000; 342(25):1871-1876. doi: https://doi.org/10.1056/NEJM200006223422504
9. Faglia G, Arosio M, Bazzoni N. Ectopic acromegaly. Endocrinol Metab Clin North Am. 1992; 21(3):575-595
10. Guillemin R, Brazeau P, Böhlen P, et al. Growth hormone-releasing factor from a human pancreatic tumor that caused acromegaly. Science. 1982; 218(4572):585-587. doi: https://doi.org/10.1126/science.6812220
11. Rivier J, Spiess J, Thorner M, Vale W. Characterization of a growth hormone-releasing factor from a human pancreatic islet tumour. Nature. 1982; 300(5889):276-278. doi: https://doi.org/10.1038/300276a0
12. Garby L, Caron P, Claustrat F, et al. Clinical characteristics and outcome of acromegaly induced by ectopic secretion of growth hormone-releasing hormone (GHRH): a French nationwide series of 21 cases. J Clin Endocrinol Metab. 2012; 97(6):2093-2104. doi: https://doi.org/10.1210/jc.2011-2930
13. Ghazi AA, Amirbaigloo A, Dezfooli AA, et al. Ectopic acromegaly due to growth hormone releasing hormone. Endocrine. 2013; 43(2):293-302. doi: https://doi.org/10.1007/s12020-012-9790-0
14. Zendran I, Gut G, Kałużny M, et al. Acromegaly caused by ectopic growth hormone releasing hormone secretion: a review. Front Endocrinol (Lausanne). 2022; 13:867965. doi: https://doi.org/10.3389/fendo.2022.867965
15. Ravindra VM, Raheja A, Corn H, et al. Primary pituitary diffuse large B-cell lymphoma with somatotroph hyperplasia and acromegaly: case report. J Neurosurg. 2017; 126(5):1725-1730. doi: https://doi.org/10.3171/2016.5.JNS16828
16. Southgate HJ, Archbold GP, el-Sayed ME, et al. Ectopic release of GHRH and ACTH from an adenoid cystic carcinoma resulting in acromegaly and complicated by pituitary infarction. Postgrad Med J. 1988; 64(748):145-148. doi: https://doi.org/10.1136/pgmj.64.748.145
17. Potorac I, Bonneville JF, Daly AF, et al. Pituitary MRI Features in Acromegaly Resulting From Ectopic GHRH Secretion From a Neuroendocrine Tumor: Analysis of 30 Cases [published correction appears in J Clin Endocrinol Metab. 2022 Nov 23;107(11):e4332]. J Clin Endocrinol Metab. 2022;107(8):e3313-e3320. doi: https://doi.org/10.1210/clinem/dgac274
18. Lonser RR, Kindzelski BA, Mehta GU, et al. Acromegaly without imaging evidence of pituitary adenoma. J Clin Endocrinol Metab. 2010; 95(9):4192-4196. doi: https://doi.org/10.1210/jc.2010-0570
19. Khandelwal D, Khadgawat R, Mukund A, Suri A. Acromegaly with no pituitary adenoma and no evidence of ectopic source. Indian J Endocrinol Metab. 2011; 15 Suppl 3(Suppl3):S250-S252. doi: https://doi.org/10.4103/2230-8210.84878
20. Scheithauer BW, Carpenter PC, Bloch B, Brazeau P. Ectopic secretion of a growth hormone-releasing factor. Report of a case of acromegaly with bronchial carcinoid tumor. Am J Med. 1984;76(4):605-616. doi: https://doi.org/10.1016/0002-9343(84)90284-5
21. Gola M, Doga M, Bonadonna S, et al. Neuroendocrine tumors secreting growth hormone-releasing hormone: Pathophysiological and clinical aspects. Pituitary. 2006; 9(3):221-229. doi: https://doi.org/10.1007/s11102-006-0267-0
22. Bertherat J, Turpin G, Rauch C, et al. Presence of somatostatin receptors negatively coupled to adenylate cyclase in ectopic growth hormone-releasing hormone- and alpha-subunit-secreting tumors from acromegalic patients responsive to octreotide. J Clin Endocrinol Metab. 1994; 79(5):1457-1464. doi: https://doi.org/10.1210/jcem.79.5.7962343
23. Shintani Y, Yoshimoto K, Horie H, et al. Two different pituitary adenomas in a patient with multiple endocrine neoplasia type 1 associated with growth hormone-releasing hormone-producing pancreatic tumor: clinical and genetic features. Endocr J. 1995; 42(3):331-340. doi: https://doi.org/10.1507/endocrj.42.331
24. Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012; 97(9):2990-3011. doi: https://doi.org/10.1210/jc.2012-1230
25. Mamedova EO, Mokrysheva NG, Przhiialkovskaia EG, et al. Multiple endocrine neoplasia type 1 variants and phenocopies. Terapevticheskii Arkhiv. 2014; 86(10):87-91. (In Russ.)
26. Knyazeva OV. Prediktory i chastota razvitiya novoobrazovanij shchitovidnoj zhelezy i zheludochno-kishechnogo trakta u pacientov s akromegaliej [dissertation] Moscow; 2016. (In Russ.) Доступно по: https://www.endocrincentr.ru/sites/default/files/specialists/science/dissertation/1diss_kniazeva_v2.pdf?ysclid=lky09llvkf123399757 Ссылка активна на 10.08.2023 г.
Elizaveta O. Mamedova - MD, PhD.
11 Dm. Ulyanova street, 117036 Moscow
None
Elena G. Przhiyalkovskaya - MD, PhD.
Moscow
None
Svetlana A. Buryakina - MD, PhD.
Moscow
None
Ekaterina V. Bondarenko - MD, PhD.
Moscow
None
Anastasiya M. Lapshina - MD, PhD.
Moscow
None
Mikhail Yu. Pikunov - MD, PhD.
Moscow
None
Zhanna E. Belaya - MD, PhD.
Moscow
None
Galina A. Melnichenko - MD, PhD, Professor.
Moscow
None
|
|
1. Figure 1. Patient S.N.Yu. Typical changes in appearance with a mild form of acromegaly in old age: slight enlargement of facial features (A), enlargement of hands (B) | |
| Subject | ||
| Type | Исследовательские инструменты | |
View
(388KB)
|
Indexing metadata ▾ | |
|
|
2. Figure 2. MRI of the pituitary gland of patient S.N.Yu., coronal projection: A. T1 WI with contrast enhancement. Microadenoma in the left part of the adenohypophysis (arrow) B. T1 WI with contrast enhancement. Rathke's pouch cyst (arrows) B. T2 VI. Diffusely reduced MR signal on T2 WI from adenohypophysis tissue (arrow) | |
| Subject | ||
| Type | Исследовательские инструменты | |
View
(430KB)
|
Indexing metadata ▾ | |
|
|
3. Figure 3. Immunohistochemical study of lung carcinoid from patient S.N.Yu. (A) - diffuse expression of GHRH; (B) - lack of GH expression. Histoscans | |
| Subject | ||
| Type | Исследовательские инструменты | |
View
(697KB)
|
Indexing metadata ▾ | |
|
|
4. Figure 4. MRI and CT examination of patient D.I.E. a) MRI of the pituitary gland, coronal projection, T1 VI with contrast enhancement. Rathke's pouch cyst (arrow). An area of slightly reduced contrast agent accumulation in the left part of the adenohypophysis (long arrow); b) Rathke's pouch cyst (arrow). Diffusely reduced MR signal on T2 WI from adenohypophysis tissue; c) MSCT of the lungs, pulmonary mode, native phase, axial projection. Neuroendocrine tumor in S10 of the left lung (short arrow); d) MSCT of the lungs, pulmonary mode, native phase, axial projection. The mass in S3 of the left lung with intra- and peribronchial spread is the focus of screening (thick arrow). | |
| Subject | ||
| Type | Исследовательские инструменты | |
View
(397KB)
|
Indexing metadata ▾ | |
|
|
5. Figure 5. Histological and immunohistochemical examination of surgical material from the pituitary gland of patient D.I.E. (A) - adenohypophysis tissue with extensive fields of oxyphilic cells (hemotoxylin-eosin). (B) - preserved network of reticulin fibers with dilated acini (impregnation with silver). (B) - diffuse staining of cells of the adenohypophysis with GH (immunohistochemical reaction with antibodies to GH). Histoscans. | |
| Subject | ||
| Type | Исследовательские инструменты | |
View
(1MB)
|
Indexing metadata ▾ | |
|
|
6. Figure 6. Immunohistochemical study of lung carcinoid from patient D.I.E. a) diffuse expression of GHRH; b) lack of expression of growth hormone; c) positive expression of somatostatin type 2A receptors (SSTR 2A) (3 points). Histoscans | |
| Subject | ||
| Type | Исследовательские инструменты | |
View
(1MB)
|
Indexing metadata ▾ | |
|
|
7. Figure 7. MRI and CT examination of patient S.N.A. a) MRI of the pituitary gland before surgery, coronal projection, T1 VI with contrast enhancement. Pituitary macroadenoma (arrow); b) MRI of the pituitary gland after surgery, coronal projection, T1 VI with contrast enhancement. Pituitary macroadenoma (arrow), decrease in size; c) MSCT of the lungs, pulmonary mode, arterial phase, axial projection. Neuroendocrine tumor in S4 of the left lung (arrow). | |
| Subject | ||
| Type | Исследовательские инструменты | |
View
(404KB)
|
Indexing metadata ▾ | |
|
|
8. Figure 8. Histological and immunohistochemical examination of the surgical material of the pituitary tumor of patient S.N.A. a) tumor of a solid structure made of chromophobe cells (hematoxylin-eosin); b) uneven cytoplasmic staining with low molecular weight cytokeratin, clone CAM 5.2 (immunohistochemical reaction); c) diffuse pronounced expression of growth hormone in tumor cells (immunohistochemical reaction); d) pronounced expression of somatostatin 2A receptors type 12 according to IRS (immunohistochemical reaction). Histoscans. | |
| Subject | ||
| Type | Исследовательские инструменты | |
View
(1MB)
|
Indexing metadata ▾ | |
|
|
9. Figure 9. Immunohistochemical study of lung carcinoid from patient S.N.A. Diffuse expression of GHRH. UV 40. | |
| Subject | ||
| Type | Исследовательские инструменты | |
View
(642KB)
|
Indexing metadata ▾ | |
Mamedova E.O., Przhiyalkovskaya E.G., Buryakina S.A., Bondarenko E.V., Lapshina A.M., Pikunov M.Yu., Belaya Zh.E., Melnichenko G.A. Ectopic acromegaly due to bronchial neuroendocrine tumors: the first description in Russia of three clinical cases. Problems of Endocrinology. 2024;70(1):66-80. https://doi.org/10.14341/probl13346

 |
![]()
![]()

![]()
11, Dm. Ul’yanova str., Moscow, Russian Federation, 117292
Processing of personal data


































