



Contents
Scroll to:
 P. M. Khandaeva,
A. M. Lapshina,
E. G. Przhialkovskaya,
Zh. E. Belaya,
А. Yu. Grigoriev,
G. A. Mel’nichenko
P. M. Khandaeva,
A. M. Lapshina,
E. G. Przhialkovskaya,
Zh. E. Belaya,
А. Yu. Grigoriev,
G. A. Mel’nichenko https://doi.org/10.14341/probl13349
Scroll to:
According to numerous studies, the most common pituitary tumors are prolactinomas, reaching 60% of all clinically significant adenomas, the next in order are non-functional pituitary adenomas, somatotropinomas, corticotropinomas and thyrotropinomas. Plurigormonal tumors occur in less than 1% of all pituitary adenomas. The most common form of mixed secretion adenoma in this patient population, derived from the Pit-1 cell line, produces various combinations of hormones: growth hormone (GH), prolactin (PRL), thyroid-stimulating hormone (TSH). This article presents a patient with a plurihormonal two-component pituitary macroadenoma with a rare and exceptional combination of secreted hormones — GH / adrenocorticotropic hormone (ACTH) / TSH / follicle-stimulating hormone (FSH) / luteinizing hormone (LH) with minimal nonspecific clinical manifestations such as diabetes mellitus and poorly controlled arterial hypertension.
Kostyleva D.N., Khandaeva P.M., Lapshina A.M., Przhialkovskaya E.G., Belaya Zh.E., Grigoriev А.Yu., Mel’nichenko G.A. Clinical case of plurihormonal pituitary adenoma (STH/ACTH/TSH/FSH/LH-secreting), diagnostic pitfalls. Problems of Endocrinology. 2024;70(4):24-31. https://doi.org/10.14341/probl13349
Pituitary tumours account for up to 16% of all primary brain neoplasms and are the third most common intracranial tumour after glioma and meningioma in the adult population [1].
Epidemiological studies show an increase in the incidence of pituitary adenomas from 3.9 to 7.4 cases per 100,000 population per annum and prevalence from 76 to 116 cases per 100,000 in the general population, which may be due to the increased availability of imaging examination techniques. Pituitary adenomas are increasingly being detected incidentally during MRI for reasons unrelated to the adenoma (ENT diseases, neurological diseases, or craniocerebral trauma).
Among the subtypes of neuroendocrine tumours in anterior pituitary, prolactinomas are the most frequently diagnosed clinically significant ones, accounting for 40%–60% of the total, according to numerous epidemiological studies. They are followed in descending order by non-functioning pituitary adenomas (30%), somatotropinomas (10%–15%), corticotropinomas (4.0%–15%), and thyrotropinomas (0.3%–2%). Less than 1% of cases are plurihormonal pituitary adenomas; double pituitary adenomas account for 0.4%–1.3% of all cases [1][2][3][4].
According to the World Health Organisation (WHO) recommendations, adenomas should be considered as plurihormonal if they secrete more than one anterior pituitary hormone (except GH+PRL, FSH+LH secreting adenomas, which are considered monomorphic) [5]. Low detection rate of plurihormonal pituitary adenomas may be due to the relatively recent introduction of immunohistochemical analysis of removed chiasmal-sellar tumours that measures specific transcription factors and hormonal expression; frequent absence of hypersecretion of anterior pituitary hormones; and poor clinical manifestations in cases where hypersecretion is detected.
Immunophenotype analysis has demonstrated that plurihormonality is frequently found in both hormonally active and clinically non-functioning pituitary adenomas. A study by Pawlikowski M. et al. showed that more than one third (36.1%) of examined adenomas express more than one hormone, which is most often not accompanied by hypersecretion of pituitary hormones. In Ruoyu Shi et al., functionally active plurihormonal adenomas are usually clinically manifested through expression of a single hormone or are asymptomatic. Currently, data on frequency and clinically significant manifestations of plurihormonal pituitary adenomas are still scarce and contradictory [1][5][6].
According to the classification of endocrine tumours, pituitary neuroendocrine tumours (PitNETs), formerly known as pituitary adenomas, are typed using pituitary transcription factors (Pit-1, TPIT, SF1, GATA3, ER-alpha), pituitary hormones and cytokeratins. The new classification uses correlation of morphological and immunohistochemical data with the clinical course of pituitary neuroendocrine neoplasms [7].
The most common form of adenomas with mixed secretion originates from a single Pit-1 cell line; such adenomas secrete various combinations of GH, PRL, and TSH. However, there are true plurihormonal adenomas with unusual hormonal combinations that are associated with different cell lineages of differentiation. The Pit-1 family is the most complex of all and concerns the differentiation of somatotropic, lactotrophs, mammosomatotroph, thyrotropic cells. Transcription factors such as SF-1, ERa are required for the development of gonadotrophic cells and corresponding tumours; GATA-2, Lhn4, as well as TPIT and NeuroD1 for corticotrophs also giving rise to densely granulated and sparsely granulated corticotrophic tumours. Availability of immunohistochemical analysis of specific transcription factors enables to clarify cell lines of differentiation in rare plurihormonal adenomas, as it was done in this case [7].
This article presents clinical observation of a patient with a rare two-component plurihormonal pituitary adenoma (STG/ ACTH/ TTG/ FSH/ LH-secreting) with minimum clinical manifestations. Adenomas with a similar type of hormone secretion, clinical picture and features of the tumour morphological structure have not been previously described in the literature.
A male patient K., 65, visited a neurologist at his local clinic with complaints of general fatigue, shakiness, unsteadiness of gait, swelling, and impaired coordination of movements which had been disturbing him for three years. Brain MRI revealed a 14×12×17 mm intrasellar mass with clear irregular contours. It was deemed a pituitary incidentaloma, and the patient was referred to Russia’s Endocrinology Research Centre for examination and determination of further treatment.
Physical examination revealed enlarged hands (Figure 1) and no other acromegaloid changes in appearance. The patient himself did not notice any changes in appearance, nor any increase in the size of feet and hands. There were no clinical signs of hypercortisolism. Testicles were correctly shaped, of normal size, without any visual pathology. Body weight – 77.0 kg, height – 174 cm, body mass index – 25.4 kg/m². Family history was not aggravated.
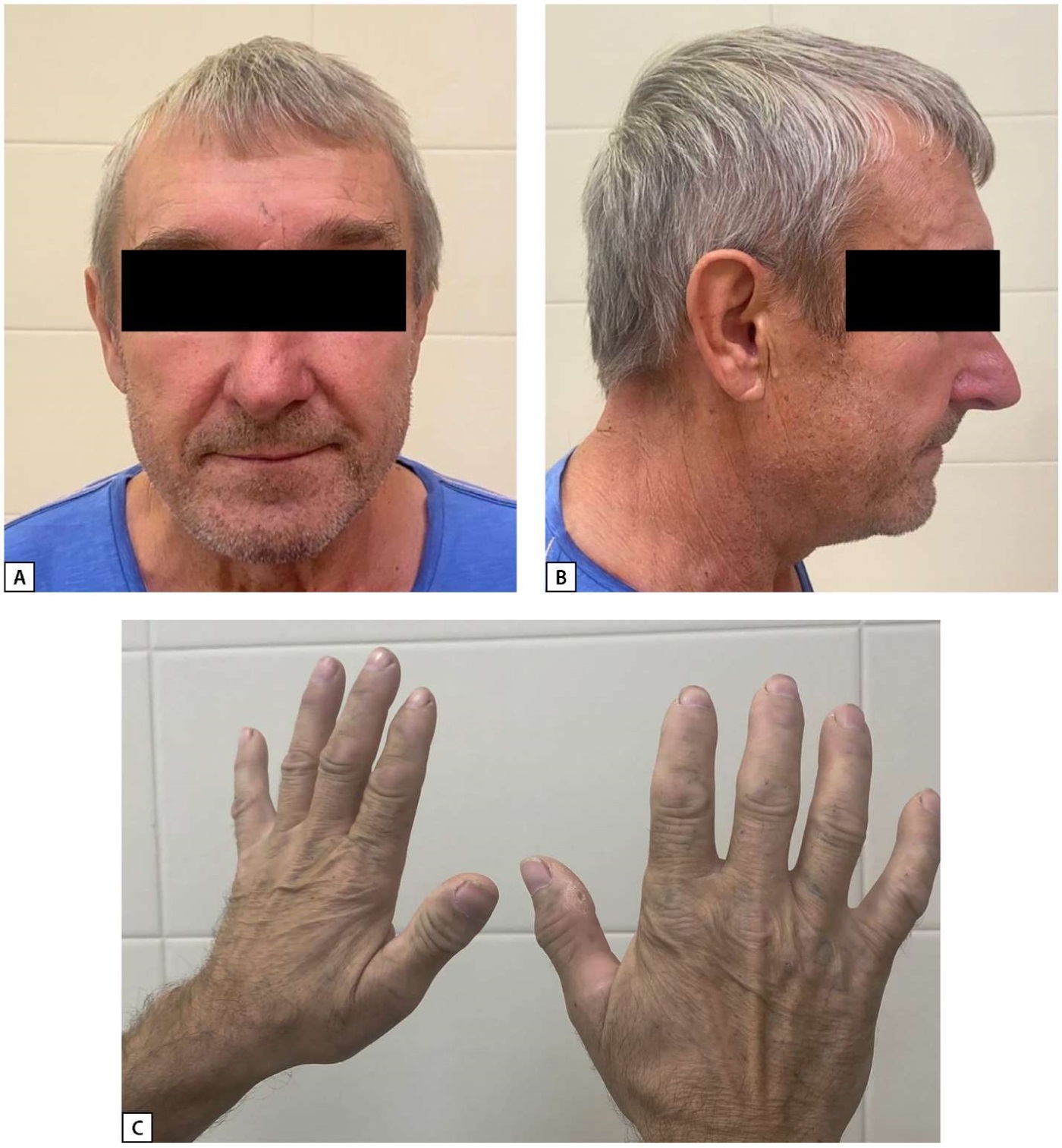
Figure 1: The patient’s appearance.
Clinical and laboratory examination revealed multiple hypersecretion of pituitary hormones. Blood hormonal examination is presented in Table 1.
Table 1: Hormonal examination results prior to and after surgical treatment
|
Before adenomectomy |
Six months after adenomectomy |
Unit of measurement |
Normal range |
|
|
HbA1c |
6.1 |
5.4 |
% |
4–6 |
|
Free cortisol in evening saliva |
14.7 |
13.3 |
nmol/L |
0.5–9.65 |
|
Evening cortisol |
622.8 |
179.8 |
nmol/L |
64–327 |
|
Morning ACTH |
37.15 |
15.18 |
pg/ml |
7.2–63.3 |
|
Morning cortisol |
680.4 |
453.1 |
nmol/L |
171–536 |
|
TSH |
0.99 |
0.525 |
mIU/L |
0.25–3.5 |
|
Free T4 |
25.21 |
12.24 |
pmol/L |
9–19 |
|
Free T3 |
5.61 |
2.73 |
pmol/L |
2.6–5.7 |
|
FSH |
- |
4.86 |
IU/L |
1.6–9.7 |
|
LH |
- |
1.86 |
IU/L |
2.5–11 |
|
Testosterone |
9.27 |
13.9 |
nmol/L |
11–28.2 |
|
PRL |
112 |
201.2 |
mIU/L |
60–355 |
|
IGF-1 |
488.7 |
192.1 |
ng/ml |
16–245 |
|
GH |
1.89 |
2.17 |
ng/ml |
0.02–1.23 |
|
Parathormone |
24.42 |
43.7 |
pg/ml |
15–65 |
|
Morning cortisol during overnight 1 mg dexamethasone suppression test |
680.4 |
49.26 |
nmol/L |
Under 50 |
|
Free cortisol in 24-h urine |
279 |
451.2 |
nmol/day |
100–379 |
|
SHBG |
72.75 |
- |
nmol/L |
20.6–76.7 |
Due to elevated free thyroxine (free T4) (25.2 pmol/L) and reference level of TSH (0.9 mU/L), therapy with somatostatin analogues was initiated. During the period of administration of 300 mg/day short-acting octreotide, within three days the level of free T4 normalised and the level of TSH significantly decreased, which is typical for TSH-secreting pituitary adenoma. Therapy with long-acting somatostatin analogues was continued in order to prepare the patient for surgical treatment to achieve stable euthyroidism. Results of the study are presented in Table 2. Also, given an elevated GH and insulin-like growth factor-1 (IGF-1), glucose loading test was performed (GH during oral glucose tolerance test); no GH suppression was achieved, which points to an active stage of acromegaly (Table 3). At the same time, there was an increase in evening serum cortisol and saliva cortisol, and no cortisol suppression during overnight 1 mg dexamethasone suppression test while 24-h urinary cortisol stayed within the reference range.
Thus, a 13×20.5×18 mm pituitary tumour with laboratory-verified plurihormonal secretion (GH/ ACTH/ TSH-secreting) with minimum clinical manifestations (dia betes and arterial hypertension, AH) was confirmed. Ophthalmological examination did not reveal evidence of chiasmal syndrome.
Given the laboratory evidence of hormonal hypersecretion, in-patient examination for complications of the underlying disease was performed. A measurement of carbohydrate metabolism confirmed diabetes first detected in 2021. During the blood sugar-controlling therapy (10 mg dapagliflozin), despite the target level of glycated haemoglobin (HbA1c) at 6.1%, the patient’s glycaemic profile showed a variability of glycaemia from 4.9 to 15.2 mmol/L during the in-patient period. Due to detected ketonuria associated with dapagliflozin administration, blood sugar-controlling therapy was corrected: the patient was switched to basal-bolus insulin therapy. During the corrected therapy, a decrease in glycaemia 24-hour variability was achieved and a trend towards target glycaemia values developed (the maximum glycaemia surge amounted to 8.5 mmol/L). 3rd-degree AH was confirmed (with achievement of optimal blood pressure control through a multicomponent drug therapy (BPA, BMAC and diuretic)), 2nd degree with high risk of adverse cardiovascular events.
Table 2: Thyroid hormone measurement results before and after a somatostatin analogue test
|
|
Prior to the test |
Three days after test initiation |
Unit of measurement |
Normal range |
|
TSH |
0.99 |
0.138< |
mlU/L |
0.25-3.5 |
|
Free T4 |
25.21 |
20.7 |
pmol/L |
9-19 |
|
Free T3 |
5.61 |
- |
pmol/L |
2.6-5.7 |
Table 3: GH analyses results during oral glucose tolerance test
|
Prior to surgical treatment |
After surgical treatment |
Unit of measurement |
|
|
GH, 0 min |
3.07 |
0.29 |
ng/ml |
|
GH, 30 min after test |
1.74 |
0.33 |
ng/ml |
|
GH, 60 min after test |
3.65 |
0.68 |
ng/ml |
|
GH, 90 min after test |
2.17 |
1.09 |
ng/ml |
|
GH, 120 min after test |
1.99 |
0.60 |
ng/ml |
Moreover, a thyroid ultrasound examination revealed echographic evidence of a bilateral multinodular goitre (EU-TIRADS2) with focal changes, secondary to an autoimmune thyroid disorder. An X-ray densitometry found no evidence of decreased bone mineral density.
For the plurihormonal pituitary adenoma, transnasal transsphenoidal adenomectomy was recommended.
A transnasal transsphenoidal adenomectomy was performed on 16 June 2022. During the surgery and the Turkish saddle revision, a tumour of varying density with petrifications was detected. In the anterior part of the Turkish saddle, a tumour of moderately dense structure (not aspirated by standard suction) had petrified fragments. The mass was removed using tumour cutters and curettes. The specimen of the removed tumour was labelled as Specimen 1. Then the second part of the tumour was removed, which was grey in colour, soft in structure and located in the posterior part of the saddle – Specimen 2.
In pathomorphological examination, a pituitary tumour of different structure was analysed. Specimen 1 contained a pituitary tumour of solid structure consisting of cells with oxyphilic and chromophobe cytoplasm (no mitoses were detected) with extensive areas of calcification. Specimen 2 contained a tumour of predominantly chromophobe cells forming numerous perivascular pseudorosettes; no mitoses were detected. An immunohistochemical study found Specimen 1 to contain diffusely located tumour cells positive to ACTH, growth hormone and TSH, and the cells nuclei were also expressing the corresponding transcription factors (TPIT, Pit-1). No expression of prolactin, LH or FSH, nor of estrogen receptor alpha (ERα) or SF1 was detected. Specimen 2 showed a different immunophenotype: the tumour cells were positive to LH, FSH and SF1 without any expression of ACTH, growth hormone, prolactin, TSH or transcription factors (TPIT, Pit-1, ERα) corresponding to the indicated cell differentiation. A slight difference in the levels of proliferative activity according to the Ki-67 index is noteworthy: in Specimen 1 the index was 1.8%, and in Specimen 2 it was 2.8%. Tumour from two samples was immunopositive to low molecular weight cytokeratin (CAM 5.2). Thus, the morphological picture was interpreted as a true plurihormonal pituitary adenoma where one component was comprised of corticotropic, thyro- and somatotrophic cells with lower proliferative activity. The second component was comprised of gonadotrophic cells with higher proliferative activity (Figure 2).
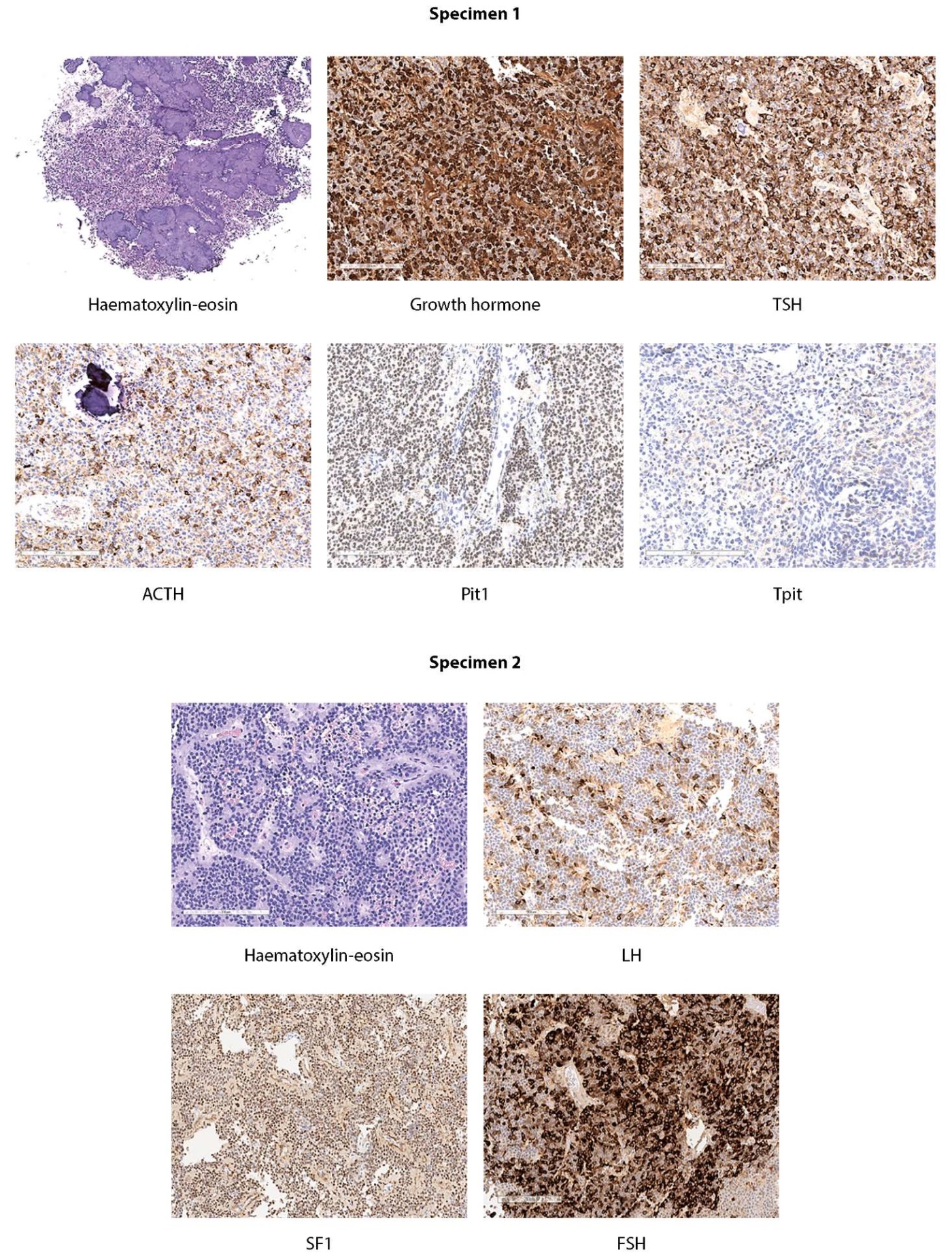
Figure 2: Microscopic structure of tumour from two specimens of removed tissue.
Plurihormonal pituitary adenoma, one of the components of which is represented by corticotrophic, thyrotrophic and somatotrophic cells with less proliferative activity. The second component — gonadotrophic cells with higher proliferative activity.
The postoperative period was normal, without any complications; the patient reported improvement of his general condition, regression of general fatigue, gait unsteadiness, swelling of the face and lower extremities. Laboratory control provided discordant data regarding persistence of endogenous hypercortisolism: there was an increase of cortisol in evening saliva (12.9 nmol/L (0.5–9.65) with normalisation of its level in 24-h urine (366 nmol/day (100–379)). We also observed GH suppression during OGTT to 0.2 ng/ml (Table 3) and decreased levels of TSH – 0.005 mIU/L (0.25–3.5) with normal levels of free T3 and free T4, which confirms achievement of postoperative remission from acromegaly and central thyrotoxicosis.
After surgical treatment, due to normalisation of blood pressure, antihypertensive therapy was cancelled; given the target glycaemic values, insulin therapy was cancelled, and the patient was switched to oral sugar-controlling therapy: 500 mg metformin twice a day, 50 mg vildagliptin twice a day, and since the start of this treatment a target glycaemic profile has been observed.
In October 2022, 6 months after neurosurgical treatment, the patient was hospitalised for a repeat in-patient evaluation. Clinical and laboratory examination provided data indicating normalisation of growth hormone and TSH secretion with persistence of discordant parameters related to central hypercortisolism: the 24-hour rhythm of ACTH secretion was disturbed; there was an increase of cortisol in evening saliva and 24-h urine, with normal evening blood cortisol and positive overnight 1 mg dexamethasone suppression test. Hormonal blood test data are presented in Table 2. A contrast brain MRI was performed, and the MR picture corresponds to a postoperative cystic fibrosis lesion (a 16×13×9 mm area of altered signal in the central and left sections of anterior pituitary). The patient was seen by a neurosurgeon; at present, there are no indications for a repeated surgical treatment, and regular follow-up has been prescribed.
Diagnosis of plurihormonal pituitary adenomas is challenging at the initial stage. The main difficulty in delivering a timely diagnosis may arise from the absence of a vivid clinical picture despite the presence of hypersecretion of pituitary hormones [6]. For example, in the clinical case described by Felix I. et al., a patient had a double pituitary macroadenoma positive for GH, TSH and a-subunit, as per an immunohistochemical examination, with elevated TSH and reference values of free T3 and free T4 in serum; there were no signs of central thyrotoxicosis, and the manifestations of acromegaly were not pronounced clinically and biochemically [8]. In a comparative study conducted by Wang M. et al., which included 279 patients with somatomammotropinomas or pure GH and PRL-secreting adenomas, it was observed that clinical manifestations of acromegaly were sparser in patients with mixed adenomas, and cardiovascular and metabolic disorders were likewise not pronounced [9]. Another recently published systematic review of the literature analysed 21 cases of plurihormonal pituitary adenomas with mixed secretion of ACTH and GH, of which two cases had signs of hypercortisolism, five had both acromegaly and hypercortisolism, and another eleven had signs of acromegaly. Interestingly, in six cases, in addition to ACTH and GH, PRL secretion was observed [10].
In plurihormonal pituitary adenomas, clinical picture of ACTH overproduction is rare and occurs with an incidence of 3.6% [11]. This suggests that the hormones secreted by the tumour are not always biologically active or lose their activity on entering the bloodstream. Also of interest is the possibility of transformation of a hormone-secreting pituitary adenoma from one type of secretion to another, which is more often observed in multiple endocrine neoplasia syndromes [12].
Determination of the biological activity of hormones secreted by tumours of this type has not been previously investigated and may be a promising area of laboratory diagnostics.
It is not uncommon for plurihormonal adenomas to debut with diabetes or AH, which is confirmed by the clinical case described here and by several others [13]. It is worth noting that plurihormonal adenomas are prone to an aggressive course, especially in case of ACTH co-secretion due to the high relapse rate [5][14]. In the clinical case presented here, the patient still has elevated cortisol in evening saliva and in 24-h urine while normal evening blood cortisol and positive overnight 1 mg dexamethasone suppression test, which may indicate persistence of endogenous hypercortisolism. However, given the lack of data on residual adenoma tissue on MRI and clinical manifestations of the disease, regular follow-up has been recommended.
The literature also shows that in patients with mixed secretion of hormones of different differentiation cell lines, e.g. ACTH, TSH, GH and FSH/LH, in most cases such pituitary adenomas secrete two hormones [6][15].
It is worth noting that in this clinical case, two components with different morphological and immunohistochemical characteristics were visually observed during surgical treatment, which may well suggest a double pituitary adenoma, where one part of the tumour produces ACTH, TSH and GH, and the other produces FSH and LH; such pituitary adenomas have not been previously described in the literature.
This clinical case demonstrates the importance of immunohistochemical study of postoperative material, which enables clarifying the nature of tumour secretion and establish its heterogeneity, thus verifying the diagnosis and determining the further course of treatment and follow-up.
We have offered the first-ever description of a clinical case where a patient has a pituitary macroadenoma with an exceptional combination of hormone secretion (GH, ACTH, TSH, FSH and LH) and has no severe manifestations of acromegaly, Cushing’s disease or central hyperthyroidism.
This observation report highlights the need to introduce immunohistochemical examinations into routine practice so as to determine not only the expression of all hormones recommended by WHO, but also the expression of transcription factors, which can increase the frequency of detection of plurihormonal pituitary adenomas. The fact that pituitary hormone secretion is not correlated with such hormones serum levels nor with the severity of clinical syndromes may provide an incentive for the development of new diagnostic methods.
Source of funding. Data collection and analysis and manuscript preparation were funded by the Russian Science Foundation (Grant 19-15-00398).
Conflict of interest. The authors hereby declare they have had no apparent or potential conflict of interest related to the content of this publication.
Authors’ contribution. Every author has approved the final version of the text prior to publication and agreed to accept responsibility for all aspects of this study, which implies due investigation and resolution of any issue related to the accuracy or integrity of any part thereof.
Patient’s consent. The patient has voluntarily given his informed consent for the publication of his personal medical information in anonymised form.
1. La Rosa S, Uccella S. Pituitary Tumors: Pathology and Genetics. In: Reference Module in Biomedical Sciences. Elsevier; 2018. doi: https://doi.org/10.1016/B978-0-12-801238-3.65086-9
2. Daly AF, Beckers A. The Epidemiology of Pituitary Adenomas. Endocrinol Metab Clin North Am. 2020;49(3):347-355. doi: https://doi.org/10.1016/j.ecl.2020.04.002
3. Irfan H, Shafiq W, Siddiqi AI, et al. Prolactinoma: Clinical Characteristics, Management and Outcome. Cureus. 2022;14(10):e29822. doi: https://doi.org/10.7759/cureus.29822
4. Eremkina AK, Dzeranova LK, Pigarova EK, Mokrysheva NG, Dedov II. Morfofunktsional’nye osobennosti gormonal’no-neaktivnykh adenom gipofiza [Morphofunctional features of non-functioning pituitary adenomas]. Arkh Patol. 2019;81(1):71-78. doi: https://doi.org/10.17116/patol20198101171
5. Pawlikowski M, Kunert-Radek J, Radek M. Plurihormonality of pituitary adenomas in light of immunohistochemical studies. Endokrynol Pol. 2010;61(1):63-66
6. Shi R, Wan X, Yan Z, Tan Z, Liu X, Lei T. Clinicopathological Characteristics of Plurihormonal Pituitary Adenoma. Front Surg. 2022;9:826720. doi: https://doi.org/10.3389/fsurg.2022.826720
7. Asa SL, Mete O, Perry A, Osamura RY. Overview of the 2022 WHO Classification of Pituitary Tumors. Endocr Pathol. 2022;33(1):6-26. doi: https://doi.org/10.1007/s12022-022-09703-7
8. Felix I, Asa SL, Kovacs K, Horvath E, Smyth HS. Recurrent plurihormonal bimorphous pituitary adenoma producing growth hormone, thyrotropin, and prolactin. Arch Pathol Lab Med. 1994;118(1):66-70
9. Wang M, Mou C, Jiang M, et al. The characteristics of acromegalic patients with hyperprolactinemia and the differences in patients with merely GH-secreting adenomas: clinical analysis of 279 cases. Eur J Endocrinol. 2012;166(5):797-802. doi: https://doi.org/10.1530/EJE-11-1119
10. Roca E, Mattogno PP, Porcelli T, Poliani L, et al. Plurihormonal ACTH-GH Pituitary Adenoma: Case Report and Systematic Literature Review. World Neurosurg. 2018;114:e158-e164. doi: https://doi.org/10.1016/j.wneu.2018.02.120
11. Allehaibi E, AlMalki MH, Brema I. Plurihormonal pituitary macroadenoma: a case report. J Med Case Rep. 2021;15(1):407. doi: https://doi.org/10.1186/s13256-021-02948-6
12. Rozhinskaya LY, Khandaeva PM, Lutsenko AS, et al. Relapse of the pituitary adenoma with a change of its hormonal activity in a female patient with multiple endocrine neoplasia syndrome type 1. Almanac of Clinical Medicine. 2018;46(3):270-275. (in Russ.) doi: https://doi.org/10.18786/2072-0505-2018-46-3-270-275
13. Tretyak OE, Shchekaturova LV, Kidalova GA, Kogut DG. Vpervye vyyavlennyj saharnyj diabet kak manifestaciya plyurigormonal’noj opuholi gipofiza (somatotropinomy- kortikotropinomy). Klinicheskij sluchaj. 2016;4(56). (In Russ.).] URL: https://cyberleninka.ru/article/n/vpervye-vyyavlennyy-saharnyy-diabet-kak-manifestatsiya-plyurigormonalnoy-opuholi-gipofiza-somatotropinomy-kortikotropinomy (дата обращения: 21.08.2023)
14. Bradley KJ, Wass JA, Turner HE. Non-functioning pituitary adenomas with positive immunoreactivity for ACTH behave more aggressively than ACTH immunonegative tumours but do not recur more frequently. Clin Endocrinol (Oxf). 2003;58(1):59-64. doi: https://doi.org/10.1046/j.1365-2265.2003.01674.x
15. Zieliński G, Maksymowicz M, Podgórski J, Olszewski WT. Double, synchronous pituitary adenomas causing acromegaly and Cushing’s disease. A case report and review of literature. Endocr Pathol. 2013;24(2):92-99. doi: https://doi.org/10.1007/s12022-013-9237-z
Darya N. Kostyleva
Moscow
none
Patimat M. Khandaeva - MD, PhD.
11 Dm. Ulyanova street, 117036 Moscow
none
Anastasia M. Lapshina - MD, PhD.
Moscow
none
Elena G. Przhiyalkovskaya - MD, PhD.
Moscow
none
Zhanna E. Belaya - MD, PhD, Professor.
Moscow
none
Andrey Yu. Grigoriev - PhD, MD.
Moscow
none
Galina A. Melnichenko - MD, PhD, professor, acad.
Moscow
none
|
|
1. Figure 1. Patient's appearance. | |
| Subject | ||
| Type | Исследовательские инструменты | |
View
(577KB)
|
Indexing metadata ▾ | |
|
|
2. Figure 2. Microscopic structure of tumor from different samples of removed tissue. | |
| Subject | ||
| Type | Исследовательские инструменты | |
View
(2MB)
|
Indexing metadata ▾ | |
Kostyleva D.N., Khandaeva P.M., Lapshina A.M., Przhialkovskaya E.G., Belaya Zh.E., Grigoriev А.Yu., Mel’nichenko G.A. Clinical case of plurihormonal pituitary adenoma (STH/ACTH/TSH/FSH/LH-secreting), diagnostic pitfalls. Problems of Endocrinology. 2024;70(4):24-31. https://doi.org/10.14341/probl13349

 |
![]()
![]()

![]()
11, Dm. Ul’yanova str., Moscow, Russian Federation, 117292
Processing of personal data



































