



Содержание
Перейти к:
 А. А. Буянова,
И. Г. Воронцова,
А. Ф. Самитова,
Ю. А. Василиадис,
Е. Е. Петряйкина,
Е. С. Демина,
А. Н. Тюльпаков
А. А. Буянова,
И. Г. Воронцова,
А. Ф. Самитова,
Ю. А. Василиадис,
Е. Е. Петряйкина,
Е. С. Демина,
А. Н. Тюльпаков https://doi.org/10.14341/probl13436
Перейти к:
Термин нарушения формирования пола (НФП) объединяет группу врожденных состояний, при которых имеет место несоответствие между хромосомным и (или) гонадным полом и строением половых органов. К одной из групп НФП относятся тестикулярные нарушения при кариотипе 46,XX (ТНФП_46,XX), в структуре которой выделяют формы, обусловленные транслокацией гена SRY, и более редко — SRY-негативные формы. В настоящем сообщении нами представлено наблюдение пациента с SRY-негативным ТНФП_46,XX, у которого первоначально состояние расценивалось как вирильная форма врожденной дисфункции коры надпочечников (ВДКН), затем — как идиопатическая внутриутробная вирилизация у девочки. На фоне вирилизации в возрасте 11 лет было заподозрено наличие тестикулярной ткани. При молекулярно-генетическом обследовании (полноэкзомное секвенирование с валидацией методом С энгера) был обнаружен de novo вариант в экзоне 9 гена WT1 (chr11:32413528T>C), который по предсказаниям не приводил к изменению аминокислотной последовательности (p.Thr479=, NM_024426.6), однако нарушал сплайсинг, результатом чего было характерное для ТНФП_46,XX изменение C-концевого домена WT1. После верификации диагноза проведена гонадэктомия и назначена заместительная терапия эстрогенами. Таким образом, нами описан пациент с редкой формой ТНФП_46,XX, обусловленного вариантом в гене WT1. Представленное наблюдение иллюстрирует сложности дифференциальной диагностики синдрома внутриутробной вирилизации при женском кариотипе.
Буянова А.А., Воронцова И.Г., Самитова А.Ф., Василиадис Ю.А., Петряйкина Е.Е., Демина Е.С., Тюльпаков А.Н. Случай тестикулярного нарушения формирования пола при кариотипе 46,XX, обусловленный мнимым синонимичным вариантом в гене WT1: трудности дифференциальной диагностики синдрома внутриутробной вирилизации у девочки. Проблемы Эндокринологии. 2025;71(1):60-65. https://doi.org/10.14341/probl13436
Buianova A.A., Vorontsova I.G., Samitova A.F., Vasiliadis Yu.A., Petryaykina E.E., Demina E.S., Tyulpakov A.N. A case of 46,XX testicular disorders of sex development due to an apparent synonymous variant in the WT1 gene: difficulties of differential diagnosis of intrauterine virililzation syndrome in a girl. Problems of Endocrinology. 2025;71(1):60-65. (In Russ.) https://doi.org/10.14341/probl13436
Термин нарушения формирования пола (НФП) объединяет группу врожденных состояний, при которых имеет место несоответствие между хромосомным и (или) гонадным полом и строением половых органов [1]. У млекопитающих дифференцировка пола в период внутриутробного развития является чрезвычайно сложным процессом, затрагивающим поэтапное включение разнообразных транскрипционных факторов, оказывающих как активирующее, так и супрессирующее воздействие. У человека до 6-й недели гестации гонада бипотенциальна, и ее дальнейшая дифференцировка в яичко или яичник зависит от хромосомного пола и происходит на фоне конкурентного взаимодействия так называемых протестикулярных и проовариальных факторов [2]. Ведущая роль в дифференцировке яичка принадлежит транскрипционному фактору SRY. Данный белок, кодируемый одноименным геном на коротком плече Y-хромосомы в локусе Yp11.2, активирует транскрипцию генов группы SOXE, прежде всего SOX9, что запускает процессы, инициирующие формирование яичка, а также оказывает репрессивное воздействие на проовариальное развитие [3]. В отсутствие SRY активируются несколько сигнальных путей с участием проовариальных генов, таких как WNT4/RSPO1, FOXL2 и RUNX1 [3].
Следует отметить, что наличие кариотипа 46,XX полностью не исключает закладку тестикулярной ткани. В структуре НФП 46,XX выделяют группу тестикулярных и овотестикулярных нарушений, когда при «женском» кариотипе происходит дифференцировка гонад в яичко или овотестис соответственно [4]. Нередко при таких состояниях выявляется транслокация гена SRY, как правило на хромосому X, однако у части пациентов ген SRY отсутствует. Механизм закладки яичка при SRY-негативных формах тестикулярного НФП 46,XX (ТНФП_46,XX) не всегда очевиден. В ряде случаев он может быть объяснен повышенной экспрессией генов, которые в норме активируются SRY, что наблюдается, например, при дупликациях в регуляторной области SOX9 [5]. Между тем причиной ТНФП_46,XX могут быть и патогенные варианты в других генах, отличных от генов группы SOXE. К их числу относятся описанные недавно варианты, затрагивающие консервативный C-концевой домен транскрипционного фактора WT1 [6][7].
Нами представлено описание клинического случая, первоначально расцениваемого как вирильная форма ВДКН, а затем как идиопатическая внутриутробная вирилизация у девочки. Диагноз был уточнен при проведении полноэкзомного секвенирования, при котором был выявлен вариант в гене WT1 с типичной локализацией для ТНФП_46,XX.
Ребенок от третьей беременности. Роды 2-е срочные, вес — 3460 г, рост — 53 см. Неправильное строение наружных гениталий выявлено при рождении. На 7-е сутки переведена в стационар для обследования, где на основании проявлений вирилизации, данных кариотипа (46,XX) и повышения уровня тестостерона (4,9 нмоль/л) установлен диагноз: ВДКН, вирильная форма. Однако, учитывая отсутствие электролитных нарушений и нормальный уровень 17OHP в крови (при скрининге — 3,8 нмоль/л, ретест — 8,6 нмоль/л), от терапии глюкокортикоидами было решено воздержаться.
При стационарном обследовании в возрасте 10 мес была констатирована III ст. вирилизации по Прадеру. ДНК-анализ на ген SRY был отрицательный. При гормональном обследовании: АКТГ — 16,6 пг/мл (0–46), 17OHP — 0,7 нмоль/л (0,1–2,9), ренин — 5,1 нг/мл/ч (1,9–6,0), кортизол — 159 нмоль/л (130–640), ЛГ — 0,3 Ед/л (0,1–3,9), ФСГ — 4,7 Ед/л (0,6–6,1), тестостерон базальный <0,35 нмоль/л (0,1–0,4), тестостерон на 3-дневной пробе с хорионическим гонадотропином (ХГ) — 3,7 нмоль/л. Полученные результаты позволили исключить ВДКН. Предварительный диагноз: «Дисгенезия гонад? Идиопатическая внутриутробная вирилизация?» В возрасте 1,5 года выполнен 1-й этап феминизирующей пластики. В последующем наблюдалась амбулаторно. При контрольных стационарных обследованиях состояние без существенной динамики.
Повторное обращение в возрасте 11 лет 9 мес в связи с жалобами на увеличение клитора, огрубление голоса. При поступлении: рост — 150 см (SDS роста: 0,35), вес — 44 кг (SDS ИМТ: +0,63). В соматическом статусе без особенностей. Половое развитие по Таннеру B1P2. Наружные гениталии: гипертрофия головки клитора, половые губы увеличены в размерах, мошонкообразные, hymen не эстрогенизирован, не гиперемирован, визуализируется вход во влагалище, сформированный оперативным путем. Костный возраст: соответствует 11,5 года. При УЗИ органов малого таза определены матка (27*8*13 мм) и яичники (правый 15*10 мм, левый 14*8 мм) без выраженного фолликулярного аппарата. При гормональном обследовании: ЛГ — 8,92 МЕ/л, (0–4,3), ФСГ — 40,07 МЕ/л (0,3–7,8), эстрадиол <36,70 пмоль/л (0–345), тестостерон — 12,76 нмоль/л (0–0,98), дегидроэпиандростерон-сульфат — 0,84 мкмоль/л (0,9–7,3), бета-ХГЧ — 0,24 МЕ/л (0–4,7), СА-125 — 8,96 Ед/ мл (0–35), альфафетопротеин — 0,54 Мед/мл (0–7,29). В качестве вероятной причины внутриутробной вирилизации рассматривался дефицит ароматазы. Для оценки состояния гипоталамо-гипофизарно-гонадной системы пациенту была назначена пробная терапия эстрогенами — эстрадиола валерат per os в дозе 1,0 мг — 1 нед., затем 2,0 мг — 1 нед. Гормональные показатели через 2 нед. терапии эстрогенами: ЛГ — 3,4 МЕ/л, ФСГ — 16,4 МЕ/л, эстрадиол — 139,7 пмоль/л, тестостерон — 1,7 нмоль/л. Динамика гормональных показателей свидетельствовала о сохранной регуляции гипоталамо-гипофизарно-гонадной системы в ответ на лечение эстрогенами.
Для уточнения диагноза проведена молекулярно-генетическая диагностика. После получения информированного согласия родителей произведено выделение геномной ДНК из образца периферической крови пациента с использованием наборов Qiagen. Пробоподготовка включала ультразвуковую фрагментацию геномной ДНК (Covaris S220) и обогащение библиотек фрагментов ДНК последовательностями экзонов с помощью зондов Agilent All Exon v8 по лабораторному протоколу [8]. Секвенирование белок-кодирующих последовательностей осуществлялось методом парно-концевых прочтений на приборе G-400 (MGI Tech) по протоколу производителя. Для анализа данных использован автоматизированный пайплайн, написанный на языке python3, включающий в себя несколько этапов. Оценку качества секвенирования проводили программой FastQC v0.11.9, разбалансированные основания в начале прочтений были удалены программой BBDuk v38.96. Выравнивание прочтений на референсную сборку генома человека GRCh37/hg19 осуществлялось при помощи bwa-mem2 v2.2.1. Дубликаты были маркированы программой Picard v2.22.4 и исключены из дальнейшего анализа. Коллинг вариантов осуществлялся с помощью bcftools mpileup v1.9 и Strelka2 v2.9.2 с получением файлов формата vcf. Они были нормализованы программой vt normalize v0.5772 и отфильтрованы по таргетным регионам, расширенным на ± 100 пар оснований с каждого конца. Метрики покрытия были получены программой Picard v2.22.4. Аннотация полученных вариантов была реализована при помощи ANNOVAR. Анализ числа копий (инсерций и делеций) проводился с помощью CNVkit, аннотация полученных вариантов числа копий в соответствии с критериями ACMG [9]. Интерпретация клинической значимости выявленных вариантов производилась в соответствии с критериями ACMG [10][11] с использованием баз данных вариантов и литературных источников. В результате ДНК-анализа у пациента обнаружен вариант нуклеотидной последовательности в экзоне 9 гена WT1 (chr11:32413528T>C) в гетерозиготном состоянии (p.Thr479=, NM_024426.6).
Праймеры, необходимые для валидации гена WT1 у родителей, а также для подтверждения найденного варианта у пациента с помощью секвенирования по Сэнгеру, были разработаны с использованием Primer3Plus. ПЦР проводили в конечном объеме 25 мкл с использованием 50x Tersus полимеразы (Evrogen), 0,5 мкл 50x dNTP, по 1 мкл каждого праймера с концентрацией 10 мкМ и 50 нг геномной ДНК. Полученные ампликоны длиной 582 п.н. очищали с помощью магнитных частиц KAPA HyperPure Beads (Roche). Секвенирование образцов методом Сэнгера проводилось на генетическом анализаторе 3500xL Applied Biosystems в компании «Евроген».
Полученные результаты подтвердили отсутствие мутации у родителей и позволили установить диагноз ТНФП_XX (рис. 1).
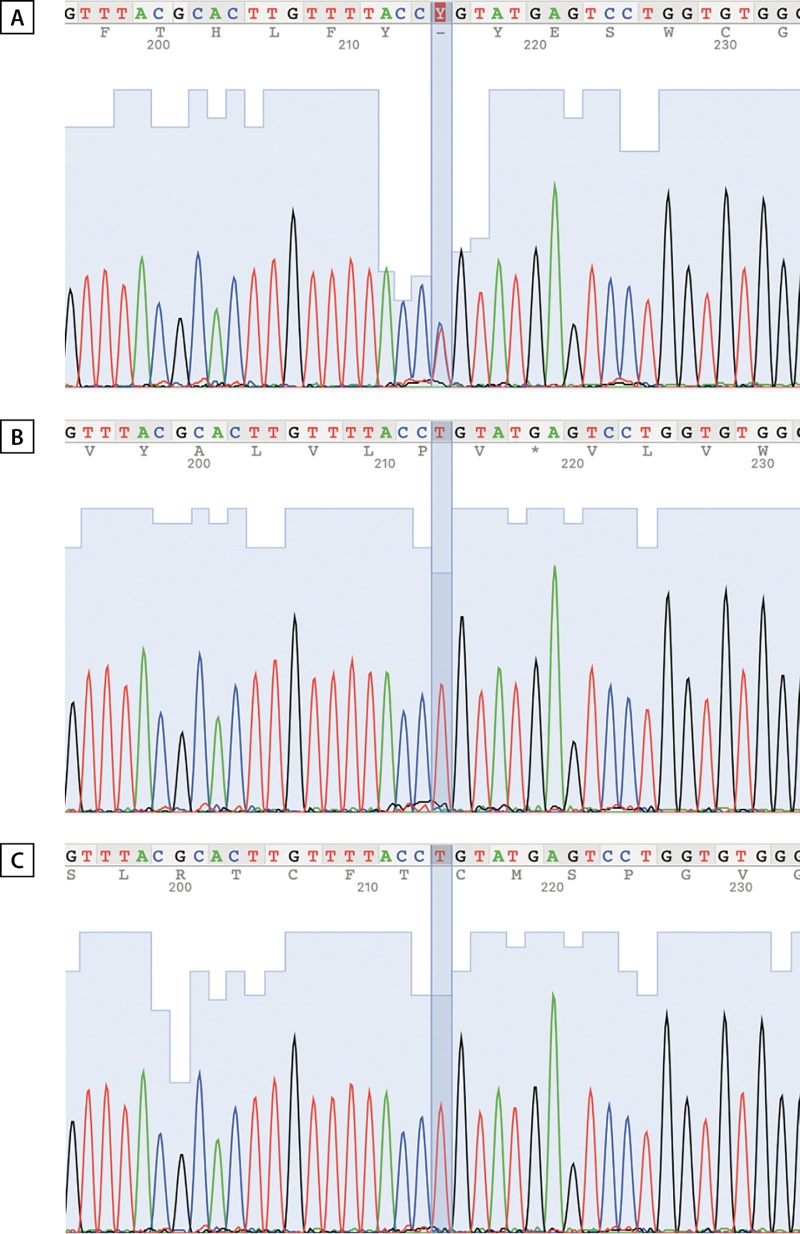
Рисунок 1. Сравнение хроматограмм последовательностей.
A — пациент, B — отец пациента, C — мать пациента.
В возрасте 11 лет 10 мес пациентке проведена операция. При лапароскопии тело матки определено по средней линии, в виде соединительнотканного тяжа, маточные трубы не определяются, в латеральных каналах за подвздошными сосудами с обеих сторон визуализированы гонады, правая — 20*10*22 мм, левая — 28*15*25 мм, серой окраски, овальной формы, поверхность гладкая, с окружающими тканями не спаяна. Произведена двусторонняя гонадэктомия. При гистологическом исследовании определена ткань гонад, представленная многочисленными тесно расположенными семенными канальцами, в которых определяются клетки Сертоли, некоторые канальцы атрофированы, гиалинизированы, между канальцами определяются клетки Лейдига. Пациентка выписана на терапии эстрогенами — эстрадиола валерат 0,5 мг/сут с последующим повышением дозы.
Представленное нами наблюдение иллюстрирует сложности дифференциальной диагностики НФП. В классификации моногенных форм НФП, предложенной европейскими экспертами, выделяют состояния с избытком андрогенов при женском кариотипе (46,XX DSD with androgen excess) [4], к которым относятся несколько форм ВДКН (гены CYP21A2, CYP11B1, HSD3B2, POR), резистентность к глюкокортикоидам (NR3C1), резистентность к эстрогенам (ESR1) и дефицит ароматазы (CYP19A1). ВДКН является, безусловно, самой частой причиной внутриутробной вирилизации у девочек, однако отсутствие повышения 17OHP в крови при нормальных уровнях кортизола и АКТГ позволили исключить данный диагноз, в том числе его редкие формы. Вирилизация и повышение уровня андрогенов при резистентности к глюкокортикоидам сопровождаются также повышением концентраций кортизола и АКТГ, что не отмечалось при обследовании пациента. Дефект рецептора к эстрогенам (ESR1) был также исключен на основании низкого уровня эстрадиола. Что касается дефицита ароматазы, то данный диагноз рассматривался нами как вероятный. В пользу дефицита CYP19A1 были внутриутробная вирилизация без прогрессии после рождения, повторное повышение уровня андрогенов в период пубертата, неопределяемые концентрации эстрадиола в пубертате и эстроген-зависимое снижение уровней тестостерона и гонадотропинов. Между тем некоторые признаки не укладывались в дефицит ароматазы, прежде всего — отсутствие увеличенных поликистозных яичников и незначительное повышение уровней гонадотропинов. Дифференциально-диагностический поиск был завершен при проведении полноэкзомного секвенирования — выявлен патогенный вариант в гене WT1, ассоциированный с ТНФП_46,XX.
Ген WT1 был локализован на коротком плече 11-й хромосомы в участке 11p13 в процессе картирования региона, делецированного при опухоли Вильмса (Wilms tumor) [12]. Белок WT1 имеет 4 основные изоформы, насчитывающие от 502 до 522 аминокислотных остатков, и представляет собой содержащий цинковые пальцы ДНК-связывающий белок (zinc finger protein), функционально являющийся транскрипционным активатором или репрессором в зависимости от тканевого или хромосомного контекста [13]. WT1 играет важную роль в процессе эмбрионального развития мочеполовой системы и мезотелиальных тканей [14], и нарушения его функции ассоциированы с широким спектром врожденной патологии почек и НФП. Герминальные мутации в гене WT1 описаны при опухоли Вильмса, тип 1 [15], синдроме Дениса-Драша (сочетание опухоли Вильмса, НФП 46,XY и гломерулонефрита) [16], синдроме Фрезье (сочетание дисгенезии гонад при кариотипе 46,XY и гломерулонефрита) [17], нефротическом синдроме, тип 4 [18], и синдроме Мичем (НФП 46,XY, врожденная диафрагмальная грыжа и возможное сочетание с удвоением влагалища, пороком сердца и легких) [19].
Патогенные варианты в гене WT1 как причина ТНФП_46,XX были описаны совсем недавно. Eozenou с соавт. при обследовании 78 пациентов с SRY-негативными тестикулярными или овотестикулярными НФП при кариотипе 46,XX выявили 7 случаев заболевания, обусловленных патогенными de novo вариантами в гене WT1 [6]. Из 4 пациентов с ТНФП_46,XX у 3 была IV степень вирилизации по Прадеру, и гонады (дисгенетичные яички) в брюшной полости, тогда как еще в 1 случае было правильное мужское строение наружных гениталий, и яички определялись в мошонке [6]. Примечательно, что все выявленные авторами патогенные варианты в гене WT1 затрагивали последовательность последнего 10-го экзона, что приводило (как было доказано in vitro), к нарушению структурной стабильности 4-го цинкового пальца (ZF4) белка WT1 [6]. Полученные экспериментальные данные позволяют предполагать, что такие нарушения функции ZF4 приводят к относительной активации сигналинга с участием протестикулярных транскрипционных факторов (в сравнении с проовариальными), что и объясняет формирование фенотипа ТНФП_46,XX в процессе внутриутробного развития [6].
Обнаруженный нами в ходе диагностического поиска вариант WT1(NM_024426.6): c.1437A>G был ранее описан Sirokha с соавт., которые диагностировали ТНФП_46,XX у пациента с двойственным строением наружных гениталий (2-я стадия по Прадеру) и двусторонним крипторхизмом [7]. Как и в представленном нами наблюдении, авторами было отмечено повышение тестостерона в ответ на стимуляцию ХГ. Особенностью замены c.1437A>G является то, что такой вариант аннотируется при первичном биоинформатическом анализе как синонимичный p.Thr479= (кодоны ACA и ACG в положении 479 соответствуют треонину) и, как следствие, может интерпретироваться как доброкачественный. Между тем геномная позиция hg19_chr11:32413528 соответствует предпоследнему нуклеотиду одного из альтернативных экзонов гена WT1, и замена c.1437A>G приводит к нарушению консервативного донорного сайта сплайсинга, результатом чего, как было доказано in vitro [7], является удержание интрона 9 в молекуле мРНК и потеря последовательности цинкового пальца ZF4, которая в норме кодируется экзоном 10. Можно отметить, что молекулярный патогенез в результате замены c.1437A>G (дисфункция ZF4) такой же, как и при описанных ранее вариантах, ассоциированных с ТНФП_46,XX [6].
Таким образом, впервые в отечественной литературе нами описан случай ТНФП_46 XX, обусловленного вариантом в гене WT1. Представленное наблюдение иллюстрирует сложности дифференциальной диагностики синдрома внутриутробной вирилизации при женском кариотипе, проведение которой требует исключения ТНФП_46,XX, в том числе SRY-негативных форм. Выявленная нами однонуклеотидная замена в гене WT1 подчеркивает также необходимость более пристального внимания к т.н. доброкачественным вариантам (в т.ч. синонимичным), которые не встречаются или встречаются крайне редко в референсных базах данных как казуативные. Часть из них может оказывать влияние на сплайсинг, что должно учитываться при выборе соответствующих программных модулей в процессе проведения биоинформатического анализа.
Источники финансирования. Работа выполнена по инициативе авторов без привлечения финансирования.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи.
Участие авторов. Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
Согласие пациента. Пациент добровольно подписал информированное согласие на публикацию персональной медицинской информации в обезличенной форме в журнале «Проблемы эндокринологии».
1. Capel B. Vertebrate sex determination: evolutionary plasticity of a fundamental switch. Nat Rev Genet. 2017 Nov;18(11):675-689. doi: 10.1038/nrg.2017.60. Epub 2017 Aug 14. PMID: 28804140.
2. Gonen N, Lovell-Badge R. The regulation of Sox9 expression in the gonad. Curr Top Dev Biol. 2019;134:223-252. doi: 10.1016/bs.ctdb.2019.01.004. Epub 2019 Feb 13. PMID: 30999977.
3. Nicol B, Grimm SA, Chalmel F, Lecluze E, Pannetier M, Pailhoux E, Dupin-De-Beyssat E, Guiguen Y, Capel B, Yao HH. RUNX1 maintains the identity of the fetal ovary through an interplay with FOXL2. Nat Commun. 2019 Nov 11;10(1):5116. doi: 10.1038/s41467-019-13060-1. PMID: 31712577; PMCID: PMC6848188.
4. Audi L, Ahmed SF, Krone N, Cools M, McElreavey K, Holterhus PM, Greenfield A, Bashamboo A, Hiort O, Wudy SA, McGowan R; The EU COST Action. GENETICS IN ENDOCRINOLOGY: Approaches to molecular genetic diagnosis in the management of differences/disorders of sex development (DSD): position paper of EU COST Action BM 1303 ‘DSDnet’. Eur J Endocrinol. 2018 Oct 1;179(4):R197-R206. doi: 10.1530/EJE-18-0256. PMID: 30299888; PMCID: PMC6182188.
5. Hyon C, Chantot-Bastaraud S, Harbuz R, Bhouri R, Perrot N, Peycelon M, Sibony M, Rojo S, Piguel X, Bilan F, Gilbert-Dussardier B, Kitzis A, McElreavey K, Siffroi JP, Bashamboo A. Refining the regulatory region upstream of SOX9 associated with 46,XX testicular disorders of Sex Development (DSD). Am J Med Genet A. 2015 Aug;167A(8):1851-8. doi: 10.1002/ajmg.a.37101. Epub 2015 Apr 21. PMID: 25900885.
6. Eozenou C, Gonen N, Touzon MS, Jorgensen A, Yatsenko SA, Fusee L, Kamel AK, Gellen B, Guercio G, Singh P, Witchel S, Berman AJ, Mainpal R, Totonchi M, Mohseni Meybodi A, Askari M, Merel-Chali T, Bignon-Topalovic J, Migale R, Costanzo M, Marino R, Ramirez P, Perez Garrido N, Berensztein E, Mekkawy MK, Schimenti JC, Bertalan R, Mazen I, McElreavey K, Belgorosky A, Lovell-Badge R, Rajkovic A, Bashamboo A. Testis formation in XX individuals resulting from novel pathogenic variants in Wilms' tumor 1 (WT1) gene. Proc Natl Acad Sci U S A. 2020 Jun 16;117(24):13680-13688. doi: 10.1073/pnas.1921676117. Epub 2020 Jun 3. PMID: 32493750; PMCID: PMC7306989.
7. Sirokha D, Gorodna O, Vitrenko Y, Zelinska N, Ploski R, Nef S, Jaruzelska J, Kusz-Zamelczyk K, Livshits L. A Novel <i>WT1</i> Mutation Identified in a 46,XX Testicular/Ovotesticular DSD Patient Results in the Retention of Intron 9. Biology (Basel). 2021 Nov 30;10(12):1248. doi: 10.3390/biology10121248. PMID: 34943163; PMCID: PMC8698877.
8. Belova V, Pavlova A, Afasizhev R, Moskalenko V, Korzhanova M, Krivoy A, Cheranev V, Nikashin B, Bulusheva I, Rebrikov D, Korostin D. System analysis of the sequencing quality of human whole exome samples on BGI NGS platform. Sci Rep. 2022 Jan 12;12(1):609. doi: 10.1038/s41598-021-04526-8. PMID: 35022470; PMCID: PMC8755732.
9. Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011 Jul;13(7):680-5. doi: 10.1097/GIM.0b013e3182217a3a. PMID: 21681106.
10. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-24. doi: 10.1038/gim.2015.30
11. Рыжкова О. П., Кардымон О. Л., Прохорчук Е. Б., и др. Руководство по интерпретации данных последовательности ДНК человека, полученных методами массового параллельного секвенирования (MPS) (редакция 2018, версия 2). Медицинская генетика, 2019, Том 18, № 2. doi.org/10.25557/2073-7998.2019.02.3-23.
12. Rose EA, Glaser T, Jones C, Smith CL, Lewis WH, Call KM, Minden M, Champagne E, Bonetta L, Yeger H, et al. Complete physical map of the WAGR region of 11p13 localizes a candidate Wilms' tumor gene. Cell. 1990 Feb 9;60(3):495-508. doi: 10.1016/0092-8674(90)90600-j. PMID: 2154334.
13. Hossain A, Saunders GF. The human sex-determining gene SRY is a direct target of WT1. J Biol Chem. 2001 May 18;276(20):16817-23. doi: 10.1074/jbc.M009056200. Epub 2001 Feb 20. PMID: 11278460.
14. Wagner KD, Wagner N, Schley G, Theres H, Scholz H. The Wilms' tumor suppressor Wt1 encodes a transcriptional activator of the class IV POU-domain factor Pou4f2 (Brn-3b). Gene. 2003 Feb 27;305(2):217-23. doi: 10.1016/s0378-1119(02)01231-3. PMID: 12609742.
15. Pelletier J, Bruening W, Li FP, Haber DA, Glaser T, Housman DE. WT1 mutations contribute to abnormal genital system development and hereditary Wilms' tumour. Nature. 1991 Oct 3;353(6343):431-4. doi: 10.1038/353431a0. PMID: 1654525.
16. Pelletier J, Bruening W, Kashtan CE, Mauer SM, Manivel JC, Striegel JE, Houghton DC, Junien C, Habib R, Fouser L, et al. Germline mutations in the Wilms' tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell. 1991 Oct 18;67(2):437-47. doi: 10.1016/0092-8674(91)90194-4. PMID: 1655284.
17. Barbaux S, Niaudet P, Gubler MC, Grünfeld JP, Jaubert F, Kuttenn F, Fékété CN, Souleyreau-Therville N, Thibaud E, Fellous M, McElreavey K. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet. 1997 Dec;17(4):467-70. doi: 10.1038/ng1297-467. PMID: 9398852.
18. Jeanpierre C, Denamur E, Henry I, Cabanis MO, Luce S, Cécille A, Elion J, Peuchmaur M, Loirat C, Niaudet P, Gubler MC, Junien C. Identification of constitutional WT1 mutations, in patients with isolated diffuse mesangial sclerosis, and analysis of genotype/phenotype correlations by use of a computerized mutation database. Am J Hum Genet. 1998 Apr;62(4):824-33. doi: 10.1086/301806. PMID: 9529364; PMCID: PMC1377045.
19. Suri M, Kelehan P, O'neill D, Vadeyar S, Grant J, Ahmed SF, Tolmie J, McCann E, Lam W, Smith S, Fitzpatrick D, Hastie ND, Reardon W. WT1 mutations in Meacham syndrome suggest a coelomic mesothelial origin of the cardiac and diaphragmatic malformations. Am J Med Genet A. 2007 Oct 1;143A(19):2312-20. doi: 10.1002/ajmg.a.31924. PMID: 17853480.
Буянова Анастасия Александровна
Москва
Конфликта интересов нет
Воронцова Инна Геннадьевна
Москва
Конфликта интересов нет
Самитова Алина Фаритовна
Москва
Конфликта интересов нет
Василиадис Юлия Андреевна
Москва
Конфликта интересов нет
Петряйкина Елена Ефимовна, д.м.н.
119571, Москва, Ленинский пр-т, д. 117, корп. 1
Конфликта интересов нет
Демина Елена Степановна
Москва
Конфликта интересов нет
Тюльпаков Анатолий Николаевич, д.м.н.
Москва
Конфликта интересов нет
|
|
1. Рисунок 1. Сравнение хроматограмм последовательностей. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(1MB)
|
Метаданные ▾ | |
Буянова А.А., Воронцова И.Г., Самитова А.Ф., Василиадис Ю.А., Петряйкина Е.Е., Демина Е.С., Тюльпаков А.Н. Случай тестикулярного нарушения формирования пола при кариотипе 46,XX, обусловленный мнимым синонимичным вариантом в гене WT1: трудности дифференциальной диагностики синдрома внутриутробной вирилизации у девочки. Проблемы Эндокринологии. 2025;71(1):60-65. https://doi.org/10.14341/probl13436
Buianova A.A., Vorontsova I.G., Samitova A.F., Vasiliadis Yu.A., Petryaykina E.E., Demina E.S., Tyulpakov A.N. A case of 46,XX testicular disorders of sex development due to an apparent synonymous variant in the WT1 gene: difficulties of differential diagnosis of intrauterine virililzation syndrome in a girl. Problems of Endocrinology. 2025;71(1):60-65. (In Russ.) https://doi.org/10.14341/probl13436

 |
![]()
![]()

![]()
117292, Российская Федерация, Москва, ул. Дм. Ульянова, д.11




































