




Содержание
Перейти к:
https://doi.org/10.14341/probl13188
Перейти к:
Врожденный нефрогенный несахарный диабет (ННД, резистентность к антидиуретическому гормону (АДГ)) — редкое наследственное заболевание, характеризующееся нечувствительностью дистальных отделов нефрона к антидиуретическому эффекту вазопрессина. Основными клиническими проявлениями заболевания являются выраженная полиурия с гипостенурией и никтурией и полидипсия. В большинстве случаев, около 90%, ННД представляет собой Х-сцепленное рецессивное заболевание, вызванное мутациями в гене рецептора вазопрессина (AVPR2). Остальные 10% случаев опосредованы инактивирующими мутациями в гене аквапорина-2 (AQP2) и имеют аутосомно-рецессивный или аутосомно-доминантный типы наследования. Зарегистрированные на сегодняшний день нуклеотидные изменения в гене AQP2 носят спорадический характер, отсутствуют данные о наличии «частых» мутаций и распространенности заболевания как среди мировой популяции, так и среди отдельных этнических групп. В данной работе впервые в мировой литературе представлено описание 12 случаев резистентности к АДГ, обусловленной новой гомозиготной мутацией p.R113C в гене AQP2, среди коренного населения Республики Бурятия.
Макрецкая Н.А., Нанзанова У.С., Хамаганова И.Р., Еремина Е.Р., Тюльпаков А.Н. Клинические и лабораторные характеристики резистентности к антидиуретическому гормону, обусловленной новой гомозиготной мутацией p.R113C в гене AQP2. Проблемы Эндокринологии. 2023;69(2):75-79. https://doi.org/10.14341/probl13188
Makretskaya N.A., Nanzanova U.S., Hamaganova I.R., Eremina E.R., Tiulpakov A.N. Clinical and laboratory characteristics of arginine vasopressin resistance, caused by a new homozygous mutation p.R113C in AQP2. Problems of Endocrinology. 2023;69(2):75-79. (In Russ.) https://doi.org/10.14341/probl13188
Международным коллективом автором в 2022 г. предложена новая концепция названий для несахарного диабета, основанная на этиопатогенезе: для центрального несахарного диабета — «дефицит вазопрессина (антидиуретического гормона (АДГ))», для нефрогенного несахарного диабета (ННД) — «резистентность к вазопрессину (АДГ)» [1]. Обновленная номенклатура призвана повысить уровень понимания патологии врачами-неэндокринологами и снизить риски необоснованного назначения десмопрессина.
Резистентность к АДГ (ННД) представляет собой гетерогенную группу заболеваний, характеризующихся нарушением резорбции воды собирательными трубочками нефронов почек. На сегодняшний день описано два наследственных варианта развития врожденного ННД: обусловленный инактивирующими мутациями в гене AVPR2 (OMIM #300538), которые приводят к нарушению чувствительности рецептора к действию вазопрессина (AVP), и в гене AQP2 (OMIM #107777), участвующем в процессе реабсорбции воды в просветах собирательных трубочек [2][3].
По данным литературы, около 90% всех наследственных форм резистентности к АДГ вызвано мутациями в гене AVPR2 с Х-сцепленным рецессивным типом наследования [4]. Остальные 10% случаев обусловлены патологическими изменениями в гене AQP2 с аутосомно-рецессивным или аутосомно-доминантным типами наследования заболевания [2][5]. К настоящему моменту описано более 70 патогенных вариантов, расположенных по всей протяженности гена AQP2, в 74 семьях (http://www.hgmd.cf.ac.uk), что свидетельствует о единичном распространении отдельных мутаций.
Известно, что в относительно изолированных этнических группах накопление отдельных мутантных аллелей приводит к распространению этноспецифических заболеваний.
В данном исследовании нами представлено описание 12 пациентов — этнических бурятов с резистентностью к АДГ, обусловленной гомозиготной мутацией p.R113C в гене AQP2.
В исследование включены 12 пациентов из 11 семей с диагнозом «нефрогенный несахарный диабет» (5 девочек, 7 мальчиков). Этническая принадлежность обследуемых — буряты. Все пациенты обследованы в Республиканской детской клинической больнице, г. Улан-Удэ.
Первоначально ННД был заподозрен у девочки 4 лет (пациент №1, табл. 1). Учитывая пол ребенка и отсутствие клинических проявлений заболевания у кого-либо из родителей, было предположено наличие рецессивного варианта ННД, и при секвенировании гена AQP2 обнаружена гомозиготная мутация c.337C>T p.R113C (рис. 1). В период с 2014 по 2020 гг. схожая клиническая симптоматика была отмечена еще у 11 пациентов той же этнической группы, которым уже прицельно проводилось исследование гена AQP2, выявлена аналогичная мутация. Таким образом, по результатам молекулярно-генетического исследования гомозиготный вариант c.337C>T p.R113C в гене AQP2 обнаружен у 12 пробандов из 11 семей (5 девочек, 7 мальчиков) (табл. 1).
Геномную ДНК выделяли из лейкоцитов периферический крови наборами PureLink® Genomic DNA Mini Kit (Thermo Scientific, Waltham, MA, USA). Применялся метод секвенирования по Сэнгеру на секвенаторе Genetic Analyzer Model 3130 (Thermo Scientific, Waltham, MA, USA) с использованием следующего набора праймеров:
AQ2_1F: GCCTTGAGAAAGAGAGCGATAG;
AQ2_1R: CAGAGCCCATCCCTCCCATCTC;
AQ2_2F: CGTCTGGCAAGCCCAGGTGTTC;
AQ2_3F: CCTTTAGGCTGAGGTCAAG;
AQ2_4R: CACGTCCAGGAAGCAGCTACTC.
Оценка патогенности варианта нуклеотидной последовательности проводилась согласно международным и российским рекомендациям [6][7]. Нумерация кодирующей последовательности гена AQP2 дана по референсу NM_000486.3 (http://www.ncbi.nlm.nih.gov/genbank). Для сравнения частоты нуклеотидного варианта использованы данные gnomAD (https://gnomad.broadinstitute.org/) [8].
Ведущими клиническими симптомами у всех обследуемых пациентов с гомозиготной мутацией p.R113C в гене AQP2 являлись полиурия и полидипсия до нескольких литров сутки (табл. 1). При сборе анамнестических данных установлено, что описанные жалобы беспокоили с раннего возраста (Ме возраста начала клинических проявлений составила 5 [ 3; 9] мес), со слов родителей, среднее количество выпиваемой жидкости к возрасту 1 года составляло 2–3 л/сут, в общем анализе мочи амбулаторно выявлено снижение относительной плотности мочи до 1000–1003 г/л. Отмечалось постепенное нарастание симптоматики с возрастом ребенка. У 3 пациентов (№ 3, 7, 10) также на первом году жизни наблюдались эпизоды субфебрилитета до 37,6°С, без признаков присоединения интеркуррентных заболеваний (за дополнительным обследованием не обращались), купировавшиеся самостоятельно на фоне дотации жидкости. Медикаментозная терапия обследуемым пациентам не назначалась.
Ме возраста постановки диагноза составила 3 [ 2,25; 4,5] года. На момент госпитализации у всех пациентов отмечалась полиурия со снижением относительной плотности мочи (Ме 1001 [ 1001; 1002] г/л). В биохимическом анализе крови в 3 случаях выявлено повышение уровня натрия (№ 5, 7, 8), Ме натрия составила 144 [ 139; 146] ммоль/л. Уровни калия (Ме 4,3 [ 4,1; 4,4] ммоль/л), глюкозы (Ме 4,3 [ 4,1; 4,7] ммоль/л) и кальция (Ме 2,5 [ 2,4; 2,6] ммоль/л) соответствовали референсным интервалам. У 7 пациентов выявлено повышение уровня осмолярности крови, Ме 301,5 [ 293; 307] мОсм/кг. По техническим причинам исследование уровня осмолярности мочи выполнено не было. По данным ультразвукового исследования почек в 6 случаях диагностирована пиелоэктазия. Диагноз «нефрогенный несахарный диабет» устанавливался на основании проведения пробы с десмопрессином (за исключением пациента №6–2): на фоне приема препарата не выявлено значимого повышения уровня относительной плотности мочи (Ме 1002 [1001–1003]). Пациенту 6–2 диагноз установлен на основании клинической картины и отягощенной наследственности по данному заболеванию.
Таблица 1. Клинические и лабораторные данные пациентов
№ | Пол | Возраст манифестации, месяцы | Возраст постановки диагноза, годы | Полиурия/ полидипсия | Диурез, мл | Относительная плотность мочи, г/л | Na сыворотки, ммоль/л | К сыворотки, ммоль/л | Глюкоза, ммоль/л | Са общий, ммоль/л | Осмолярность крови мОсм/кг | Пиелоэктазия | Субфебрилитет | Отягощенная наследственность |
1 | Жен. | 4 | 4 | да | 5320 | 1002 | 144 | 4,1 | 4,0 | 2,4 | 302 | да | нет | нет |
2 | Жен. | 2 | 3 | да | 4460 | 1001 | 139 | 4,3 | 4,6 | 2,5 | 294 | нет | нет | нет |
3 | Муж. | 10 | 2 | да | 3570 | 1003 | 143 | 4,2 | 4,1 | 2,3 | 301 | нет | да | нет |
4 | Муж. | 12 | 3 | да | 4505 | 1000 | 137 | 4,5 | 4,2 | 2,7 | 290 | нет | нет | нет |
5 | Муж. | 7 | 1,5 | да | 3780 | 1001 | 146 | 4,3 | 4,8 | 2,5 | 307 | да | нет | нет |
6–1 | Жен. | 3 | 5 | да | 5640 | 1002 | 145 | 4,0 | 4,1 | 2,3 | 304 | да | нет | да, сибс 6–2 |
6–2 | Муж. | 2 | 3 | да | 3830 | 1001 | 138 | 4,4 | 4,3 | 2,7 | 291 | нет | нет | да, сибс 6–1 |
7 | Муж. | 3 | 2 | да | 3540 | 1001 | 153 | 4,5 | 5,0 | 2,2 | 322 | да | да | нет |
8 | Жен. | 11 | 7 | да | 4580 | 1001 | 147 | 3,8 | 4,1 | 2,6 | 309 | да | нет | нет |
9 | Муж. | 6 | 4 | да | 4260 | 1003 | 143 | 3,9 | 4,2 | 2,5 | 300 | нет | нет | нет |
10 | Муж. | 8 | 5 | да | 4130 | 1002 | 138 | 4,3 | 4,4 | 2,4 | 291 | да | да | нет |
11 | Жен. | 3 | 3 | да | 3890 | 1002 | 145 | 4,4 | 4,7 | 2,6 | 306 | нет | нет | нет |
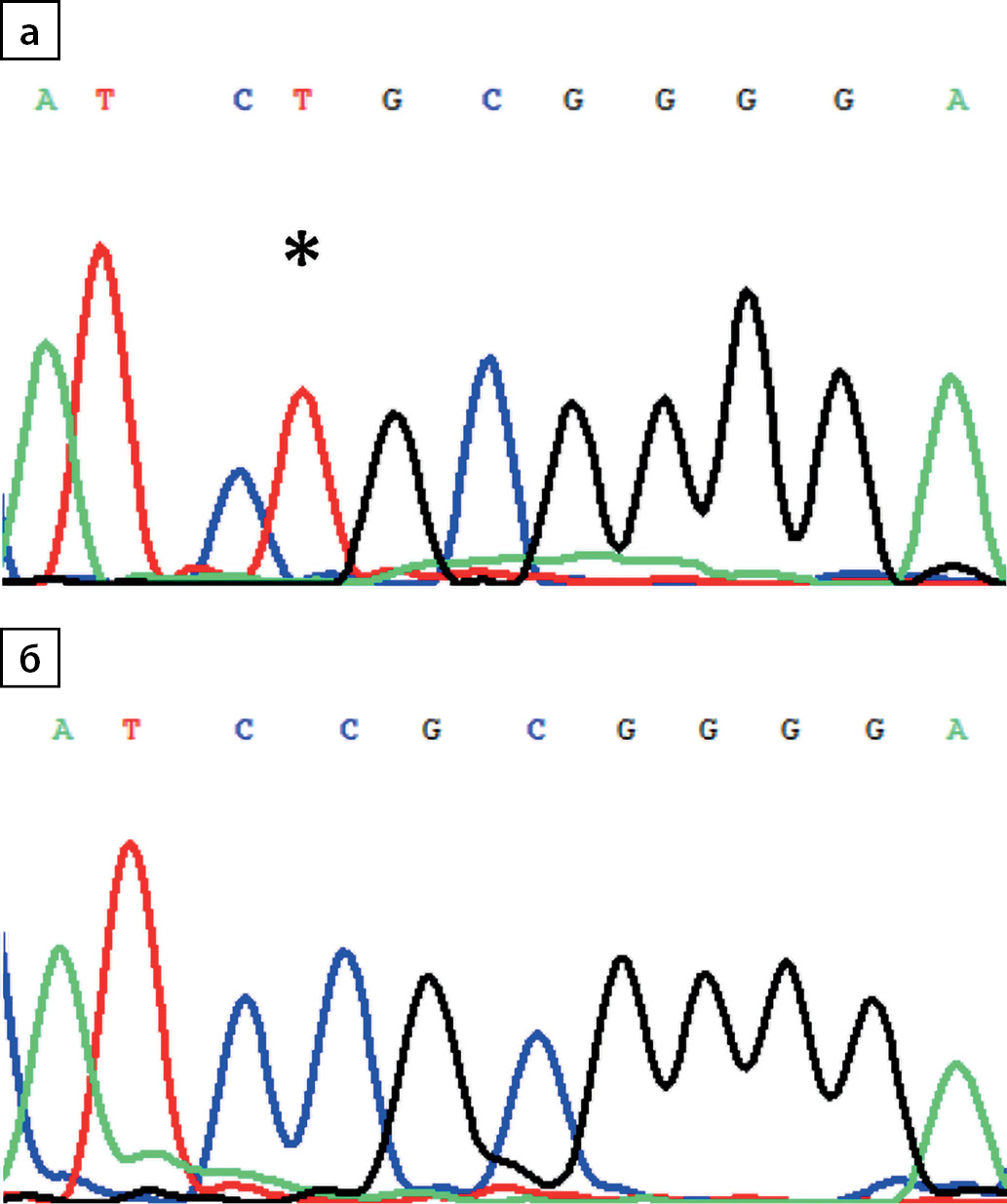
Рисунок 1. Электрофореграмма фрагмента последовательности экзона 1 гена AQP2: а) гомозиготная транзиция c.337C>T (*) с заменой кодона аргинина (CGC) на цистеин (TGC) в положении 113 (p.R113C); б) последовательность дикого типа.
Ген AQP2 (OMIM #107777) картирован на длинном плече хромосомы 12 (12q13.12) в 1993 г. (Sasaki S. и соавт.), состоит из четырех экзонов и трех интронов и кодирует белок аквапорин-2 (AQP2) [9][10].
AQP2 состоит из 271 аминокислоты с молекулярной массой 28 837 Да, в своем составе имеет N- и С-концевые домены, три экстрацеллюлярные петли, две интрацеллюлярные петли и шесть трансмембранных доменов. AQP2 регулирует реабсорбцию воды в ответ на стимуляцию AVP. При уменьшении объема плазмы или повышении ее осмоляльности в нейрогипофизе секретируется AVP и, связываясь с рецептором AVPR2 в собирательных трубочках почек, увеличивает внутриклеточную концентрацию циклического аденозинмонофосфата. Данный процесс стимулирует транспортировку AQP2 к апикальной плазматической мембране и ингибирует его интернализацию, опосредованную эндоцитозом. Как результат, происходит накопление AQP2 в апикальной плазматической мембране, что позволяет воде двигаться по осмотическому градиенту через мембрану в интерстиций, а затем в кровоток [11].
Впервые клиническая картина инактивирующих мутаций в гене AQP2 описана Р. Deen и соавт. в 1994 г. [12]. Ведущими клиническими проявлениями заболевания являются выраженная полиурия с гипостенурией и никтурией и полидипсия до 10–20 л/сут, развивающиеся на первом году жизни [5]. Кроме того, у ряда пациентов отмечается развитие неспецифических симптомов заболевания, таких как субфебрилитет, раздражительность, снижение аппетита, рвота и вялость, что может приводить к задержке роста и снижению массы тела у детей [6]. При лабораторном обследовании характерно снижение относительной плотности мочи, также возможно повышение уровня натрия и осмолярности плазмы на фоне ограничения жидкости [5][11]. У пациентов старшего возраста возможно развитие таких осложнений, как ортостатическая гипотензия, гидронефроз, мегауретер [11].
На сегодняшний день не разработано патофизиологического лечения резистентности к АДГ. Рекомендации по ведению таких пациентов направлены на восполнение потери жидкости с мочой адекватным количеством выпитой, в сочетании с низкосолевой диетой. Кроме того, возможно применение нестероидных противовоспалительных препаратов (НПВС) и диуретиков [13, 14]. НПВС, такие как ибупрофен и индометацин, улучшают способность концентрировать мочу и уменьшают ее объем на 25–50%, а комбинация с гидрохлоротиазидом оказывает аддитивный эффект [13][14]. Тиазидные диуретики эффективно снижают диурез при соблюдении диеты с очень низким содержанием натрия [13]. Калийсберегающие диуретики, такие как амилорид, могут иметь аддитивный эффект с тиазидными диуретиками через механизм ингибирования потери калия, вызванной тиазидами [14]. Однако для всех перечисленных вариантов терапии описан эффект ускользания [13][14].
Ранее в России описан клинический случай ННД, обусловленный мутацией p. D150E в гене AQP2 [15], патогенность которой была затем доказана функциональными исследованиями in vitro [16]. Выявленный в ходе настоящего исследования нуклеотидный вариант hg38_chr12:49951167C>T (c.337C>T p.R113C) приводит к замене аргинина на цистеин в положении 113. Ранее данная мутация у пациентов с резистентность к АДГ не описана. По данным базы gnomAD, общая частота для данного варианта составляет 0,00001243 (3:241 284), все 3 случая выявлены в гетерозиготном состоянии: 1 — южно-азиатская группа, 1 — восточно-азиатская группа, 1 — латиноамериканская группа [17]. Согласно критериям ACMG, обнаруженный вариант нуклеотидной последовательности может быть расценен как патогенный с уровнем значимости РМ2, РM1, PP3, PP4 [6][7]. Аргинин в 113 положении является частью консервативного интегринсвязывающего RGD-домена на второй экстрацеллюлярной петле. В исследованиях in vitro показано, что изменения в данном регионе могут приводить к нарушению процесса транспорта AQP2 к апикальной мембране клетки [18].
На сегодняшний день в мировой литературе отсутствуют данные о распространенности резистентности к АДГ, обусловленной мутациями в гене AQP2. Описанные ранее случаи данного заболевания являлись спорадическими, также не было выявлено каких-либо часто встречающихся мутаций для различных популяций. В нашем исследовании нуклеотидный вариант hg38_chr12:49951167C>T (c.337C>T p.R113C) идентифицирован в 12 случаях среди коренного населения Бурятии, что позволяет отнести данное изменение к основным причинам развития резистентности к АДГ в данной этнической группе и предположить наличие эффектов «основателя» и «бутылочного горлышка» в распространении варианта p.R113C среди коренного населения Республики Бурятия.
Впервые для мировой литературы описана клиническая характеристика резистентности к АДГ, обусловленная мутацией p.R113C в гене AQP2. Высокая распространенность данного заболевания среди коренного населения Бурятии позволит в дальнейшем проводить медико-генетическое консультирование семей в регионе, повысить настороженность врачей в отношении резистентности к АДГ, улучшит раннюю диагностику этого состояния, в том числе с использованием методов пренатальной диагностики.
Источники финансирования. Данная работа поддержана советом по грантам Президента Российской Федерации, номер гранта МК-5272.2022.3.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи.
Участие авторов. Макрецкая Н.А. — существенный вклад в дизайн исследования, сбор материала, анализ полученных данных, написание текста; Нанзанова У.С. — сбор материала, анализ полученных данных; Хамаганова И.Р. — сбор материала, анализ полученных данных; Еремина Е.Р. — сбор материала, анализ полученных данных; Тюльпаков А.Н. — концепция и дизайн исследования, внесение в рукопись существенной правки с целью повышения научной ценности статьи. Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
Согласие пациента. Добровольные информированные согласия пациентов и их законных представителей на публикацию в журнале «Проблемы эндокринологии» получены.
1. Arima H, Cheetham T, Christ-Crain M, et al. Changing the name of diabetes insipidus: a position statement of The Working Group for Renaming Diabetes Insipidus. Endocr Connect. 2022;11(11):549-552. doi: https://doi.org/10.1530/EC-22-0378
2. Bockenhauer D, Bichet DG. Pathophysiology, diagnosis and management of nephrogenic diabetes insipidus. Nat Rev Nephrol. 2015;11(10):576-588. doi: https://doi.org/10.1038/nrneph.2015.89
3. Fushimi K, Uchida S, Harat Y, et al. Cloning and expression of apical membrane water channel of rat kidney collecting tubule. Nature. 1993;361(6412):549-552. doi: https://doi.org/10.1038/361549a0
4. van den Ouweland AMW, Dreesen JCFM, Verdijk M, et al. Mutations in the vasopressin type 2 receptor gene (AVPR2) associated with nephrogenic diabetes insipidus. Nat Genet. 1992;2(2):99-102. doi: https://doi.org/10.1038/ng1092-99
5. Sands JM, Bichet DG. Nephrogenic Diabetes Insipidus. Ann Intern Med. 2006;144(3):186. doi: https://doi.org/10.7326/0003-4819-144-3-200602070-00007
6. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: https://doi.org/10.1038/gim.2015.30
7. Рыжкова О.П., Кардымон О.Л., Прохорчук Е.Б., и др. Руководство по интерпретации данных последовательности ДНК человека, полученных методами массового параллельного секвенирования (MPS) (редакция 2018, версия 2) // Медицинская генетика. — 2019. — Т. 18. — №2. — С. 3-23. doi: https://doi.org/10.25557/2073-7998.2019.02.3-23
8. Collins RL, Brand H, Karczewski KJ, et al. A structural variation reference for medical and population genetics. Nature. 2020;581(7809):444-451. doi: https://doi.org/10.1038/s41586-020-2287-8
9. Sasaki S, Fushimi K, Saito H, et al. Cloning, characterization, and chromosomal mapping of human aquaporin of collecting duct. J Clin Invest. 1994;93(3):1250-1256. doi: https://doi.org/10.1172/JCI117079
10. Sasaki S, Saito H, Saito F, et al. Cloning, expression and chromosomal mapping of human collecting duct water channel (hWCH-CD). J Am Soc Nephrol. 1993;(4):858.
11. Chen L, Higgins PJ, Zhang W. Development and diseases of the collecting duct system. Results Probl Cell Differ. 2017;(60):165-203. doi: https://doi.org/10.1007/978-3-319-51436-9_7
12. Deen PMT, Verdijk MAJ, Knoers NVAM, et al. Requirement of human renal water channel aquaporin-2 for vasopressin-dependent concentration of urine. Science (80- ). 1994;264(5155):92-95. doi: https://doi.org/10.1126/science.8140421
13. Milano S, Carmosino M, Gerbino A, et al. Hereditary nephrogenic diabetes insipidus: pathophysiology and possible treatment. An update. Int J Mol Sci. 2017;18(11):2385. doi: https://doi.org/10.3390/ijms18112385
14. Kavanagh C, Uy NS. Nephrogenic diabetes Insipidus. Pediatr Clin North Am. 2019;66(1):227-234. doi: https://doi.org/10.1016/j.pcl.2018.09.006
15. Тюльпаков А.Н., Рубцов П.М., Шандин А.Н. Семейный вариант нефрогенного несахарного диабета с частично сохранной концентрационной функцией почек, обусловленный гомозиготной мутацией D150E в гене аквапорина-2 (AQP2) // Проблемы Эндокринологии. — 2007. — Т. 53. — №5. — С. 13-18. doi: https://doi.org/10.14341/probl200753513-18
16. Guyon C, Lussier Y, Bissonnette P, et al. Characterization of D150E and G196D aquaporin-2 mutations responsible for nephrogenic diabetes insipidus: importance of a mild phenotype. Am J Physiol Renal Physiol. 2009;297(2):F489-498. doi: https://doi.org/10.1152/ajprenal.90589.2008
17. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285-291. doi: https://doi.org/10.1038/nature19057
18. Tamma G, Lasorsa D, Ranieri M, et al. Integrin signaling modulates AQP2 trafficking via Arg-Gly-Asp (RGD) motif. Cell Physiol Biochem. 2011;27(6):739-748. doi: https://doi.org/10.1159/000330082
Макрецкая Нина Алексеевна - кандидат медицинских наук.
115522, Москва, ул. Москворечье, д. 1
нет
Нанзанова Ульяна Сергеевна
Улан-Удэ
нет
Хамаганова Ирина Ремовна
Улан-Удэ
нет
Еремина Елена Робертовна - кандидат медицинских наук.
Улан-Удэ, Иркутск
нет
Тюльпаков Анатолий Николаевич - доктор медицинских наук.
Москва
нет
|
|
1. Рисунок 1. Электрофореграмма фрагмента последовательности экзона 1 гена AQP2: а) гомозиготная транзиция c.337C>T (*) с заменой кодона аргинина (CGC) на цистеин (TGC) в положении 113 (p.R113C); б) последовательность дикого типа. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(186KB)
|
Метаданные ▾ | |
Макрецкая Н.А., Нанзанова У.С., Хамаганова И.Р., Еремина Е.Р., Тюльпаков А.Н. Клинические и лабораторные характеристики резистентности к антидиуретическому гормону, обусловленной новой гомозиготной мутацией p.R113C в гене AQP2. Проблемы Эндокринологии. 2023;69(2):75-79. https://doi.org/10.14341/probl13188
Makretskaya N.A., Nanzanova U.S., Hamaganova I.R., Eremina E.R., Tiulpakov A.N. Clinical and laboratory characteristics of arginine vasopressin resistance, caused by a new homozygous mutation p.R113C in AQP2. Problems of Endocrinology. 2023;69(2):75-79. (In Russ.) https://doi.org/10.14341/probl13188

 |
![]()
![]()

![]()
117292, Российская Федерация, Москва, ул. Дм. Ульянова, д.11



































