



Содержание
Перейти к:
 Е. В. Шрёдер,
Т. А. Вадина,
Е. Н. Солодовникова,
В. В. Захарова,
М. В. Дегтярев,
М. Б. Конюхова,
Н. В. Сергеева,
О. Б. Безлепкина
Е. В. Шрёдер,
Т. А. Вадина,
Е. Н. Солодовникова,
В. В. Захарова,
М. В. Дегтярев,
М. Б. Конюхова,
Н. В. Сергеева,
О. Б. Безлепкина https://doi.org/10.14341/probl13210
Перейти к:
ОБОСНОВАНИЕ. Одна из возможных причин дисгенезии щитовидной железы (ЩЖ) при врожденном гипотиреозе (ВГ) — инактивирующие мутации в гене рецептора тиреотропного гормона (ТТГ) (TSHR) (NP_000360.2). Гетерозиготные мутации гена TSHR приводят к частичной резистентности к ТТГ, гомозиготные и компаунд-гетерозиготные — к гипоплазии ЩЖ и полной резистентности. В последние десятилетия в зарубежной литературе появляется все больше данных, касающихся изучения этой проблемы, в то время как отечественные публикации ограничены единичными исследованиями. Изучение этого вопроса необходимо для понимания этиологии, патогенеза заболевания и особенностей тактики наблюдения и лечения пациентов.
ЦЕЛЬ. Оценить частоту встречаемости патогенных вариантов гена TSHR у детей с дисгенезией ЩЖ при ВГ, изучить пути наследования заболевания в семьях и оценить фенотипические особенности.
МАТЕРИАЛЫ И МЕТОДЫ. Проведено одноцентровое интервенционное одномоментное несравнительное исследование. Обследованы дети с ВГ, обусловленным дисгенезией ЩЖ. Пациентам проведены УЗИ шеи и радиоизотопная визуализация тиреоидной ткани. Обследование проведено на фоне отмены гормональной терапии либо до ее начала. Произведена оценка структуры дисгенезии ЩЖ, выполнен поиск вариантов в гене TSHR методом NGS, обследованы родители и сибсы пациентов с мутациями гена TSHR.
РЕЗУЛЬТАТЫ. В исследование включены 95 детей (75 девочек; 20 мальчиков). Возраст на момент обследования составил 6,2 года [4,5; 8,9], медиана уровня неонатального ТТГ — 157,5 мЕ/л [60,9; 257,2]. Эктопия ЩЖ выявлена у 52% детей, аплазия — у 36%, гипоплазия и гемиагенезия — у 10 и 2% соответственно.
Мутации гена TSHR выявлены в 5,3% случаев (у 5 из 95 детей). Два ребенка имели моноаллельные варианты последовательности (по 1 гетерозиготному варианту — p.R450H и p.D487N), 3 — биаллельные (у 2 пробандов выявлена гомозиготная мутация p.S49Afs*9, 1 ребенок был компаунд-гетерозиготен по p.A485D и p.R450H). У всех пациентов по данным УЗИ имелась гипоплазия ЩЖ разной степени выраженности. Троим детям проведена тиреосцинтиграфия, выявлен сниженный захват радиофармакологического препарата (0,3–0,9%).
Обследование 15 членов семей выявило 9 человек с мутациями гена TSHR, при этом нарушений функции ЩЖ на момент обследования не было выявлено ни в одном случае.
ЗАКЛЮЧЕНИЕ. Частота мутаций гена TSHR, по нашим данным, составила 5,3%. Выявлены 2 ранее не описанные гетерозиготные мутации. Генетическое исследование может помочь с постановкой диагноза, определением дальнейшего наблюдения и генетического консультирования.
Шрёдер Е.В., Вадина Т.А., Солодовникова Е.Н., Захарова В.В., Дегтярев М.В., Конюхова М.Б., Сергеева Н.В., Безлепкина О.Б. Патогенные варианты гена TSHR у детей с дисгенезией щитовидной железы. Проблемы Эндокринологии. 2023;69(1):76-85. https://doi.org/10.14341/probl13210
Shreder E.V., Vadina T.A., Solodovnikova E.N., Zakharova V.V., Degtyarev M.V., Konyukhova M.B., Sergeeva N.V., Bezlepkina O.B. Pathogenic TSHR variants in children with thyroid dysgenesis. Problems of Endocrinology. 2023;69(1):76-85. (In Russ.) https://doi.org/10.14341/probl13210
Врожденный гипотиреоз (ВГ) у большинства пациентов обусловлен дисгенезией щитовидной железы (ЩЖ) [1–5]. В ее структуре выделяют эктопию, аплазию, гипоплазию и гемиагенезию. Дисгенезия чаще является спорадической, однако в ряде случаев идентифицируется генетическая причина [6].
Одна из возможных причин — мутация в гене рецептора ТТГ (TSHR), который принадлежит к семейству рецепторов, связанных с G-белком (гуанин-нуклеотид-связывающий белок) [6]. В литературе описаны как активирующие, так и инактивирующие мутации в гене TSHR, влияющие на способность ЩЖ синтезировать гормоны. Известно, что инактивирующие мутации вызывают резистентность к тиреотропному гормону (ТТГ) и могут приводить к ВГ [6][7]. В статье приводятся данные пациентов с ВГ, обусловленным инактивирующими мутациями TSHR.
Резистентность к ТТГ представляет собой снижение чувствительности фолликулов ЩЖ к стимуляции ТТГ вследствие генетических дефектов [7]. Гетерозиготные мутации гена TSHR приводят к частичной резистентности к ТТГ, в то время как гомозиготные и компаунд-гетерозиготные — к гипоплазии ЩЖ и полной резистентности к ТТГ [8–12].
В последние десятилетия в зарубежной литературе появляется все больше данных по изучению этой проблемы, проводится анализ частоты и клинических проявлений гипотиреоза. В 2021 г. Da D.-Z. и соавт. [13] опубликован крупный систематический обзор, касающийся изучения патогенных вариантов TSHR при ВГ, частоты их встречаемости и клинических особенностей пациентов с этим генотипом. Отечественные публикации ограничены единичными исследованиями [14–16].
Поскольку ген TSHR является одним из факторов, участвующих в развитии ВГ, обусловленного гипоплазией, его изучение в клинической практике необходимо для понимания этиологии, патогенеза заболевания и особенности тактики ведения пациентов.
Оценить частоту встречаемости патогенных вариантов гена TSHR у детей с дисгенезией ЩЖ при ВГ, изучить пути наследования заболевания в семьях и оценить фенотипические особенности.
Место и время проведения исследования
Место проведения. Обследование пациентов проведено в ФГБУ «НМИЦ эндокринологии» Минздрава России (далее — ЭНЦ).
Время исследования. В исследование включены пациенты, обследованные с ноября 2020 г. по июль 2022 г.
Изучаемые популяции
Критерии включения в исследование:
1. дети с ВГ, обусловленным дисгенезией ЩЖ от 0 до 18 лет;
2. родители и сибсы пациентов с мутациями гена TSHR;
3. подписанное информированное согласие на участие в исследовании.
Критерии исключения: отзыв согласия пациента или его законного представителя на участие в исследовании.
Дизайн исследования
Проведено одноцентровое интервенционное одномоментное несравнительное исследование. Набор пациентов в группы проводился на основании соответствия критериям включения и при отсутствии критериев исключения.
На первом этапе определена группа пациентов: в исследование включены дети с ВГ, обусловленным дисгенезией ЩЖ, которым проведены:
1.лабораторное обследование: определение уровней ТТГ, свободного тироксина (св.Т4), тиреоглобулина (ТГ);
2.инструментальная диагностика — комплексная визуализация тиреоидной ткани, сочетающая УЗИ шеи, планарную сцинтиграфию, однофотонную эмиссионную компьютерную томографию (ОФЭКТ) шеи и верхнего средостения с 99mТc-пертехнетатом;
3.молекулярно-генетическое исследование генов-кандидатов, ответственных за развитие ВГ.
Инструментальная диагностика и определение уровня ТГ проводились на фоне отмены гормональной терапии в течение 14 дней.
На втором этапе произведена оценка структуры дисгенезии ЩЖ у детей с ВГ, выполнен поиск вариантов в гене TSHR.
На третьем этапе обследованы родители и сибсы пациентов с выявленными мутациями гена TSHR, которым проведено секвенирование фрагмента ДНК по Сэнгеру при известной мутации. Членам семьи, у которых выявлены мутации этого гена, проведены УЗИ ЩЖ и определение уровней ТТГ и св.Т4.
Описание медицинского вмешательства
Лабораторное обследование — определение уровней ТТГ, св.Т4, ТГ в сыворотке крови проводилось в клинико-диагностической лаборатории ЭНЦ. Лабораторные исследования выполнены на автоматическом иммунохемилюминесцентном анализаторе ARCHITECT i2000sr (Abbott).
УЗИ проводилось врачом ультразвуковой диагностики на аппарате Voluson E8 expert (GE Healthcare) c линейным датчиком 11L. Исследование проведено с использованием цветового и энергетического допплеровского картирования.
Сцинтиграфия ЩЖ (области шеи и верхнего средостения) проводилась в отделении радионуклидной диагностики на гамма-камере ОФЭКТ Discovery NM630 с применением 99mТc-пертехнетата. Доза радиофармпрепарата (РФП) рассчитывалась индивидуально в зависимости от массы тела пациента с помощью калькулятора вводимой активности PedDose в МБк и мКи (https://www.eanm.org/publications/dosage-calculator). Раствор натрия 99mТc-пертехнетата получали путем элюирования генератора 99Мо/99mTc стерильным изотоническим раствором натрия хлорида. Получаемый из генератора РФП вводился внутривенно в процедурном кабинете. Исследование проводилось через 15–20 мин после внутривенного введения РФП на гамма-камере в положении пациента лежа на спине в режиме статического планарного снимка (10 мин) и затем в режиме ОФЭКТ (15 мин). Обработка полученных данных выполнялась на рабочей станции Xeleris (GE Healthcare) c использованием итеративных методов реконструкции данных и получением трехмерного изображения распределения РФП в тканях шеи и верхнего средостения с последующим описанием анатомо-физиологических характеристик визуализирующейся тиреоидной ткани.
Молекулярно-генетическое исследование проводилось в лаборатории генетики моногенных эндокринных заболеваний. Забор крови проводился из локтевой вены вне зависимости от приема пищи в пробирки с консервантом этилендиаминтетраацетатом в концентрации 1,2–2,0 мг на 1 мл крови. Геномную ДНК извлекали роботизированной станцией Allsheng Autopure-96 (Hangzhou Allsheng Instruments Co., Ltd., China) из периферической крови с использованием набора для выделения геномной ДНК из цельной крови NucleoMag Blood (MN). Выделенную ДНК качественно и количественно анализировали с помощью Quant-iT™ dsDNA HS Assay (Invitrogen, Carlsbad, CA, USA) и спектрофотометра Eppendorf Biospectrometer Fluorescence (Eppendorf AG, Germany) соответственно. Подготовку библиотеки ампликонов, подготовку и обогащение матрицы ДНК проводили в соответствии с протоколами производителей (Roche (La Roche Ltd)). Кастомная панель включала в том числе кодирующие области 28 генов: CACNA1C, DUOX1, DUOX2, DUOXA2, FOXE1, GLIS3, GNAS, IGSF1, IYD, KMT2D, NKX2-1, NKX2-5, PAX8, SECISBP2, SLC16A2, SLC26A4, SLC5A5, TBX1, TG, THRA, THRB, TPO, TRH, TRHR, TSHB, TSHR, TTR, UBR1, которые, по научным литературным данным и базе данных из OMIM, были описаны при гипотиреозе. Исследование проведено методом массового параллельного секвенирования (next-generation sequencing, NGS) на платформе Illumina методом парно-концевого чтения (2x150 п.о.).
Обработка данных секвенирования проведена с использованием автоматизированного алгоритма, включающего выравнивание прочтений на референсную последовательность генома человека (HG38), постпроцессинг выравнивания, выявление вариантов и фильтрацию вариантов по качеству, а также аннотацию выявленных вариантов по всем известным транскриптам каждого гена из базы RefSeq с применением компьютерных алгоритмов предсказания патогенности вариантов (SIFT, PolyPhen-2 HDIV, Polyphen-2 HVAR, PROVEAN, CADD). Для оценки популяционных частот выявленных вариантов использованы данные международного проекта gnomAD Exomes для экзонных вариантов и базы gnomAD Genomes для интронных вариантов. Для предсказания эффекта изменений в сайтах сплайсинга и прилежащих к сайту сплайсинга интронных участках использованы программы SpliceAI и AdaBoost.
Для оценки клинической релевантности выявленных вариантов использованы база данных OMIM, HGMD, специализированные базы данных по отдельным заболеваниям (при наличии) и литературные данные. Заключение о клинической значимости найденных вариантов дано с учетом рекомендаций American College of Medical Genetics and Genomics (ACMG) и российского руководства по интерпретации данных NGS. В заключение включены только варианты, имеющие возможное отношение к клиническим проявлениям у пациента. Полиморфизмы, классифицированные по различным критериям как нейтральные, не включены в заключение.
Анализировались панели, средняя глубина покрытия которых была не менее 70x, процент целевых нуклеотидов с эффективным покрытием >10х — не менее 97%.
Стоит отметить, что метод NGS не позволяет достоверно выявлять инсерции и делеции длиной более 10 п.о., мутации в интронных областях (за исключением канонических сайтов сплайсинга), а также мутации в генах, у которых в геноме существует близкий по последовательности паралог (псевдоген). Методика NGS не предназначена для определения фазы пар гетерозиготных мутаций и выявления мутаций в состоянии мозаицизма.
Для подтверждения патогенности обнаруженных вариантов в гене TSHR проведено секвенирование по Сэнгеру ДНК крови родителей и сибсов на генетическом анализаторе Applied Biosystems 3500.
Этическая экспертиза
Исследование одобрено локальным этическим комитетом, протокол №17 от 28.10.2020. Информированное согласие получено от всех обследованных пациентов. В том случае, если возраст обследованных не достиг 15 лет, информированное согласие подписано законным представителем.
В исследование включены 95 детей с ВГ, обусловленным дисгенезией ЩЖ (75 девочек; 20 мальчиков). Возраст на момент обследования составил 6,2 года [ 4,5; 8,9], медиана уровня неонатального ТТГ — 157,5 мЕ/л [ 60,9; 257,2]. Структура дисгенезии (по данным метода комплексной визуализации тиреоидной ткани) представлена на рисунке 1. Эктопия ЩЖ наблюдалась в половине случаев (52%), аплазия ЩЖ имелась у 36% пациентов, гипоплазия и гемиагенезия — у 10 и 2 пациентов соответственно.
По результатам молекулярно-генетического исследования мутации гена TSHR выявлены в 5,3% случаев (у 5 из 95 детей). Два пациента (NN1-2) имели моноаллельные варианты последовательности: пациент 1 имел гетерозиготный вариант p.R450H, а пациент 2 — гетерозиготный p.D487N. Три ребенка (N3-5) имели биаллельные мутации: у 2 пробандов (дети от близкородственного брака — родители троюродные брат и сестра) выявлена гомозиготная мутация p.S49Afs*9, 1 ребенок был компаунд-гетерозиготен по p.A485D и p.R450H. В таблице 1 представлены результаты обследования пациентов.
В неонатальном периоде 3 детей (NN1-3) имели повышение уровня ТТГ по данным скрининга от 10,8 до 15,1 Ед/л, в то время как у ребенка (N5) с гомозиготной мутацией и выраженной гипоплазией ЩЖ выявлено значительное повышение ТТГ — до 96,8 мЕд/л. У всех пациентов по данным УЗИ отмечалась гипоплазия ЩЖ разной степени выраженности. Троим детям проведена тиреосцинтиграфия, по данным которой обращал на себя внимание сниженный захват РФП (со значениями от 0,3 до 0,9%). Суточная доза левотироксина натрия у детей с гетерозиготными и компаунд-гетерозиготной мутациями была ниже, чем у детей с гомозиготной мутацией (табл. 1), что также свидетельствовало в пользу резистентности различной степени выраженности.
Для уточнения цис-/транс-положения найденных вариантов проведены секвенирование по Сэнгеру всем членам семей пробандов (NN1-5) и оценка фенотипа родственников. Обследование членов семьи (n=15) выявило еще 9 человек с мутациями гена TSHR(NP_000360.2): 3 из них были гетерозиготны по p.R450H, 1 — по p.D487N, 1 — по p.A485D, и 4 родителей из 2 семей имели по одной гетерозиготной мутации p.S49Afs*9. Для пациента N3 мы подтвердили компаунд-гетерозиготность мутации. Пациенты N4 и N5 родились от близкородственных браков (родители являются троюродными братом и сестрой). Учитывая обнаружение одинаковой мутации в этих семьях и факт их проживания в одной республике, можно предположить родственную связь этих семей. Родословная пациентов и выявленные изменения представлены на рисунке 2.
У членов семьи, являющихся носителями мутаций, определены уровни ТТГ и св.Т4, нарушений функции ЩЖ не выявлено, объем ЩЖ у родителей варьировал от 6,4 до 18,4 см3. Интересным является то, что у пациента N1 имеется дизиготный близнец 4,2 года, носитель аналогичной мутации, у которого на момент обследования уровни ТТГ и св.Т4 были в пределах нормальных значений, объем ЩЖ — 2,3 см3. Таблица 2 демонстрирует результаты обследования членов семьи.
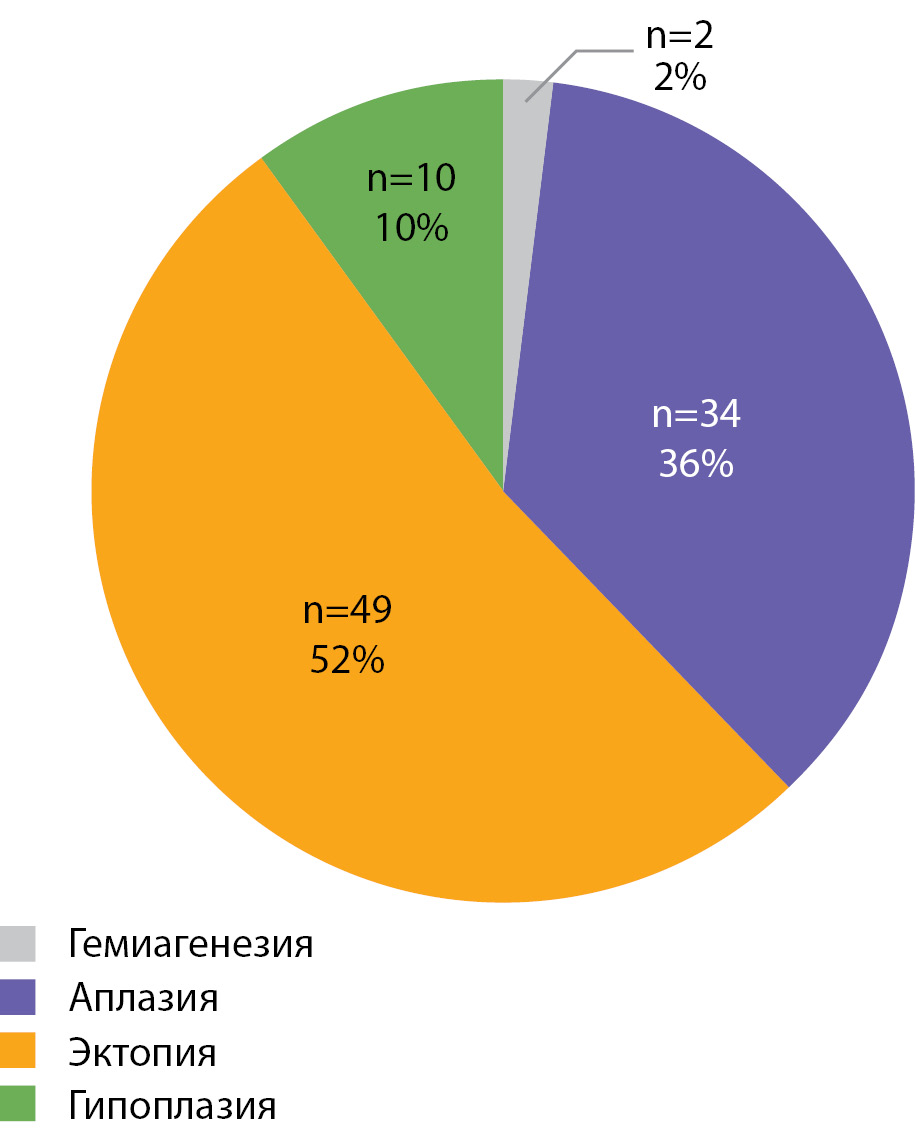
Рисунок 1. Структура дисгенезии щитовидной железы у детей с врожденным гипотиреозом.
Таблица 1. Клиническая характеристика пациентов с изменениями в гене TSHR (NM_000369.5)
Пациент | Уровень ТТГ при диагностике ВГ | Результаты обследования в ЭНЦ | Результаты | ||||||||||
ТТГ неонатальный, мЕ/л (норма до 10) | ТТГ венозной крови, мЕ/л (подтверждающая диагностика) | Возраст на момент обследования в ЭНЦ, лет | ТГ, нг/мл (норма 3,5–77) | Объем ЩЖ по данным УЗИ, см3 | Индекс захвата технеция по данным сцинтиграфии, % (норма 0,8–1,7) | Суточная доза левотироксина натрия, мкг/кг/сут | Изменение аминокислот | Изменение кДНК | Экзон | Зиготность | Классификация ACMG | Описана/ не описана | |
N1 | 10,8 | 14,2 | 4,2 | 55,5 | 1,2 | 0,3 | 1,8 | R450H | c.1349G>A | 10 | Het | Патогенный | Описана |
N2 | 12,9 | 23,4 | 4,7 | 30,7 | 1,6 | 0,5 | 2,4 | D487N | c.1459G>A | 10 | Het | Неопределенная клиническая значимость | Не описана |
N3 | 15,1 | 23,3 | 4,3 | 90,1 | 1,1 | 0,9 | 2,5 | A485D | c.1454C>A | 10 | ComHet | Вероятно патогенная | Не описана |
R450H | c.1349G>A | 10 | Вероятно патогенная | Описана | |||||||||
N4 | Нет данных | 28,4 | 7,3 | 0,9 | 0,1 | Не проведена | 3,7 | S49Afs*9 | c.144del | 1 | Homo | Патогенная | Описана |
N5 | 96,8 | 317,3 | 7,0 | 1,6 | 0,5 | Не проведена | 3,5 | S49Afs*9 | c.144del | 1 | Homo | Патогенная | Описана |
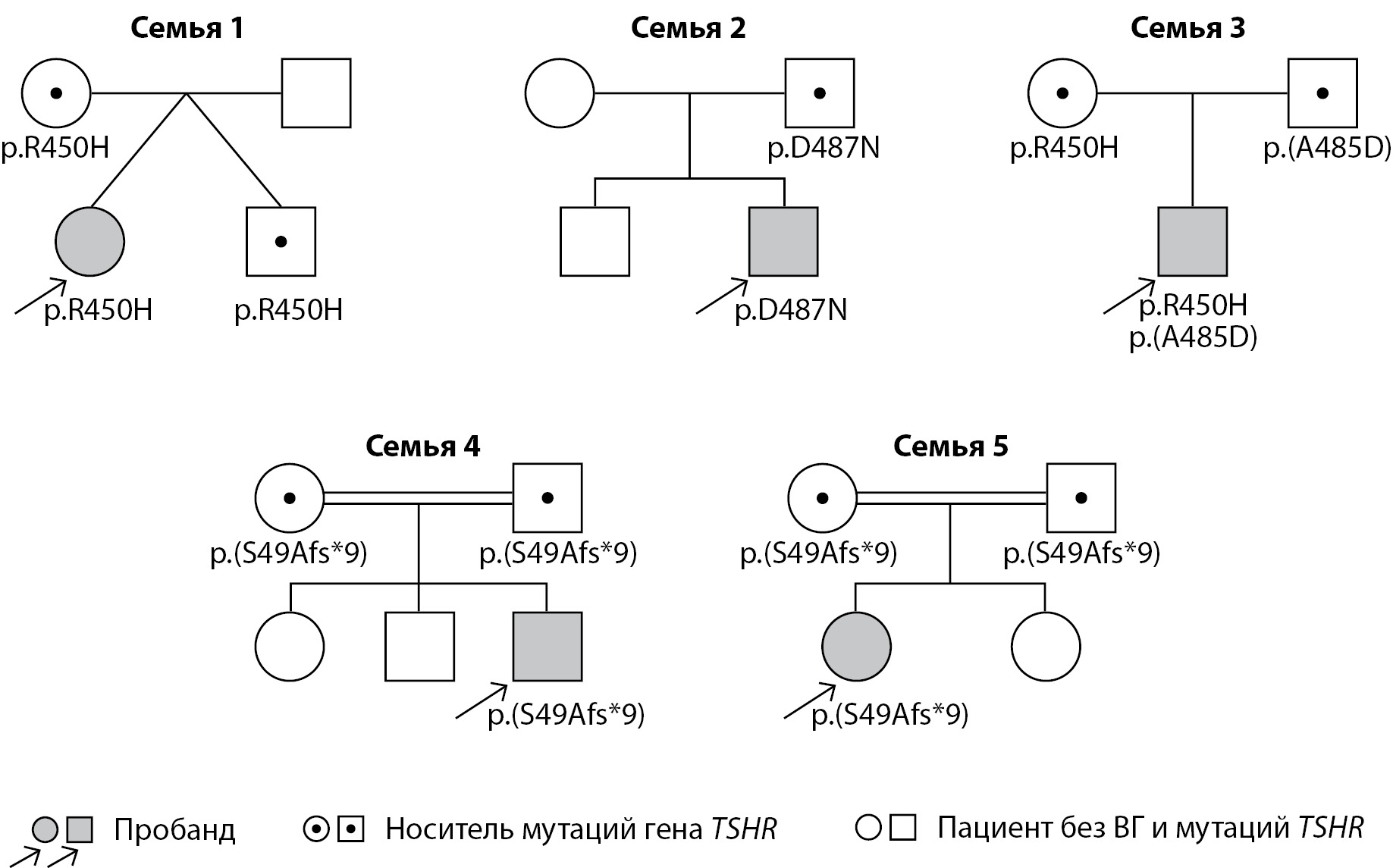
Рисунок 2. Родословные пациентов с мутацией в гене TSHR (NP_000360.2).
Таблица 2. Результаты секвенирования по Сэнгеру при известной мутации TSHR (NM_000369.5) и фенотип щитовидной железы
Пациент, N | Возраст на момент обследования, лет | Член семьи | Изменение аминокислот | Изменение кДНК | Экзон | Зиготность | Классификация ACMG | ТТГ, мЕ/л | св.Т4, пмоль/л | Объем ЩЖ, см3 |
N1 | 35 | Мать | R450H | c.1349G>A | 10 | Het | Патогенный | 2,3 | 11,4 | 16,8 |
52 | Отец | Не выявлено | Не выявлено | |||||||
4,2 | Брат | R450H | c.1349G>A | 10 | Het | Патогенный | 3,1 | 12,8 | 2,3 | |
N2 | 40 | Мать | Не выявлено | Не выявлено | ||||||
44 | Отец | D487N | c.1459G>A | 10 | Het | Неопределенная клиническая значимость | 2,2 | 12,6 | 12,3 | |
13 | Брат | Не выявлено | Не выявлено | |||||||
N3 | 40 | Мать | R450H | c.1349G>A | 10 | Het | Вероятно патогенная | Не обследована | Не обследована | Не обследована |
41 | Отец | A485D | c.1454C>A | 10 | Het | Вероятно патогенная | Не обследован | Не обследован | Не обследован | |
N4 | 32 | Мать | S49Afs*9 | c.144del | 1 | Het | Патогенный | 3,4 | 14,2 | 6,4 |
38 | Отец | S49Afs*9 | c.144del | 1 | Het | Патогенный | 2,9 | 14,2 | 18,4 | |
11 | Сестра | Не выявлено | Не выявлено | |||||||
10 | Брат | Не выявлено | Не выявлено | |||||||
N5 | 29 | Мать | S49Afs*9 | c.144del | 1 | Het | Патогенный | 2,2 | 12,9 | 9,2 |
34 | Отец | S49Afs*9 | c.144del | 1 | Het | Патогенный | 1,6 | 15 | 11,8 | |
3 | Сестра | Не выявлено | Не выявлено |
В группе обследованных детей с ВГ, обусловленным дисгенезией ЩЖ, преобладает эктопия — 52%, что согласуется с данными ранее проведенных исследований [3][4][17][18]. Второе место по частоте занимает аплазия (36%). На долю гипоплазии и гемиагенезии приходится 10 и 2% соответственно. Известно, что гипоплазия в ряде случаев обусловлена мутациями в гене TSHR [13].
Согласно систематическому обзору Da D.-Z. и соавт. 2021 г. [13], включавшему анализ данных 3975 детей с ВГ из 23 стран (представленных в 44 исследованиях «случай-контроль»), средняя частота патогенных вариантов в гене TSHR составила 7,83%. Частота патогенных вариантов в гене TSHR зависит от популяций и варьирует от 0 (Бразилия) до 29% (Израиль). Также обращает на себя внимание выраженная вариабельность в рамках одной популяции: в различных исследованиях, проведенных в Италии, она варьирует от 0 до 30,6%. Средняя частота патогенных вариантов в Европе была значительно ниже, чем в Азии.
Частота мутаций гена TSHR у пациентов с гипоплазией ЩЖ в российской популяции составила 5,3%. Наши результаты сопоставимы с результатами, полученными в Японии (Narumi S. и соавт. [19] — 5,88%), в Китае (Fang Y. и соавт. [20] — 5,91%, Ma S.G. и соавт. [21] — 5,56%, Qiu Y.L. и соавт. [22] — 5%) и Корее (Park K.J. и соавт. [23] — 5,29%). Отечественные публикации ограничены, по данным Макрецкой Н.А. и соавт. [15], частота составила 3,73%.
Изучение структуры и функции гена важно для лучшего понимания патогенеза заболевания, а данные о типе наследования и варианте мутации могут помочь в своевременной диагностике нарушений функции ЩЖ у членов семьи.
Известно, что ген TSHR впервые клонирован Parmentier М. и соавт. [24] в 1989 г. и изначально обнаружен у мышей Tshrhyt/hyt как ген, влияющий на дифференцировку ЩЖ [25].
Akamizu Т. и соавт. в 1990 г. [26] картировали ген TSHR на длинном плече 14 хромосомы человека (14q). Rousseau-Merck М. и соавт. [27] и Libert F. и соавт. [28] путем гибридизации in situ локализовали его до 14q31. Ген содержит 10 экзонов, состоит из α- и β-субъединиц, соединенных дисульфидной связью. TSHR представляет собой рецептор, связанный с G-белком, который экспрессируется на базолатеральной мембране тироцитов [29]. Основные его функции — это связывание ТТГ, регуляция роста и пролиферации клеток ЩЖ и участие в синтезе тиреоидных гормонов. ТТГ оказывает свое биологическое действие путем связывания с внеклеточным доменом рецептора ТТГ, расположенного на плазматической мембране тироцитов. Ген TSHR состоит из 7 трансмембранных доменов (transmembrane domain; TMD), соединенных через линкерную область (шарнирную область) с крупным внеклеточным доменом (extracellular domain; ECD), в основном состоящим из последовательности нескольких лейцин-богатых регионов (leucine-rich repeat regions; LRR) (рис. 3). Лейцин-богатые регионы собираются в структуру, похожую на подкову, с бета-нитями LRR, образующими вогнутую поверхность для связывания лиганда. TMD состоит из альфа-спиральных трансмембранных сегментов, соединенных внеклеточными петлями, контактирующими с лигандом ECD, и внутриклеточными петлями, участвующими в связывании G-белка [7]. Крупный ECD кодируется первыми 9 экзонами, а трансмембранные сегменты и карбоксильный конец — 10-м экзоном.
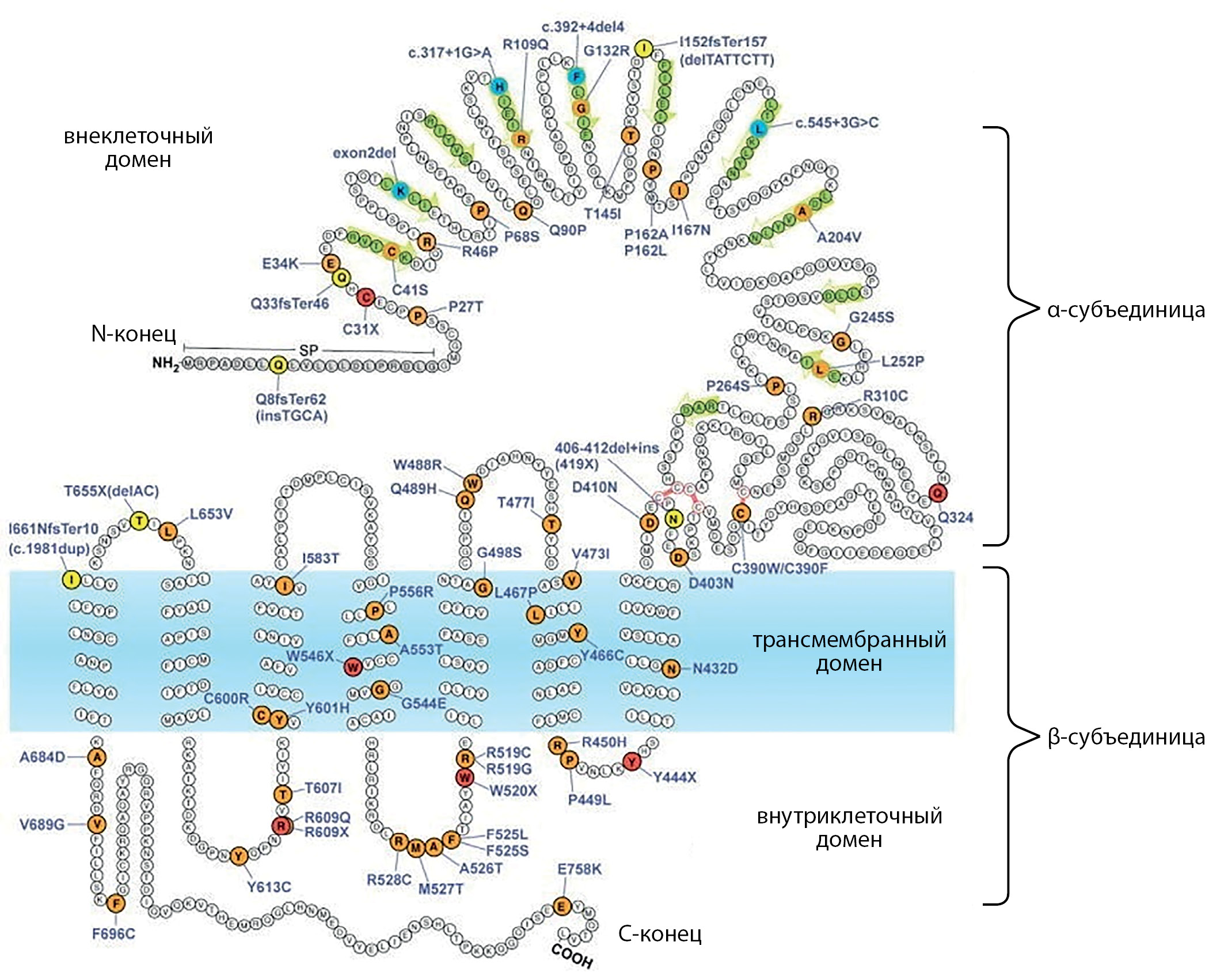
Рисунок 3. Модель TSHR с расположением подтвержденных и предполагаемых инактивирующих мутаций, выявленных у пациентов с резистентностью к ТТГ (адаптирован из [7]; с изменениями).
Инактивирующие мутации могут быть причиной развития ВГ с нарушением роста ЩЖ, приводящего к гипоплазии [7]. Для гипотиреоза, обусловленного мутациями гена TSHR, описаны как аутосомно-доминантный, так и аутосомно-рецессивный типы наследования [6–7].
Резистентность к гену TSHR зависит от зиготности и типа мутации [7][13] и варьирует от субклинического до тяжелого гипотиреоза. Выраженность резистентности к ТТГ связана с типом и локализацией мутации гена TSHR: полная потеря функции вследствие биаллельных инактивирующих мутаций обычно приводит к тяжелому ВГ с характерными клиническими проявлениями, в то время как носители других биаллельных мутаций (гомозиготные или компаунд-гетерозиготы) имеют легкую форму заболевания, проявляющуюся в виде субклинического гипотиреоза [7][30]. Пациенты с моноаллельными дефектами гена TSHR, с учетом частичной компенсации, могут не выявляться при неонатальном скрининге [31]. Таким образом, у пациентов с неаутоиммунным повышением ТТГ и низким или нормальным уровнем св.Т4 при проведении дифференциальной диагностики необходимо помнить о таком редком состоянии, как резистентность к ТТГ. В такой ситуации генетическое исследование может помочь с постановкой диагноза.
У гомозиготных носителей вариантов с потерей функции может наблюдаться выраженная гипоплазия ЩЖ, которая в ряде случаев принимается за аплазию ввиду отсутствия или снижения захвата РФП на сцинтиграфии. В этом случае определяемый уровень сывороточного ТГ позволяет дифференцировать гипоплазию от аплазии ЩЖ [7].
Таким образом, пациенты с гомозиготными вариантами с потерей функции гена TSHR нуждаются в раннем начале заместительной терапии левотироксином в высокой дозе, что демонстрируют пациенты N4 и N5. Пациенты N1–3 демонстрируют более мягкий фенотип и нуждаются в меньшей заместительной дозе препарата.
Все выявленные нами варианты у пробандов 4 и 5 представляют собой мутации с потерей функции (LOF-варианты) [7]. У 2 детей выявлен вариант p.R450H, который наиболее распространен в Азии [32][33]. Мама пациентки N1 по национальности азербайджанка, мама ребенка N3 — киргизка. Изменения у пациентов N4–5 —p.S49Afs*9 описаны лишь в российской популяции [34] и по совокупности данных являются патогенными. Две другие мутации — p.D487N и p.A485D, обнаруженные у пациентов 2 и 3, в литературе ранее не упоминались. Согласно нашим данным, с учетом фенотипа, можно предположить их участие в развитии заболевания, а следовательно, требуется дальнейшее изучение для подтверждения их патогенности.
Секвенирование гена TSHR у членов семей показало, что пациенты с двумя обнаруженными мутациями (гомозиготные и компаунд-гетерозиготные) унаследовали их от здоровых родителей, имеющих одну мутацию в гетерозиготном состоянии и нормальную функцию и объем ЩЖ. В семьях, где у пациента была найдена только одна мутация в гетерозиготном состоянии, родитель — носитель мутации не имел клинических и биохимических признаков гипотиреоза. Учитывая предполагаемый аутосомно-рецессивный тип наследования заболевания, вероятнее всего, второй генетический вариант в таких семьях не был идентифицирован ввиду ограничений метода высокопроизводительного секвенирования и требует дальнейшего поиска другими генетическими методами исследования. Целесообразно динамическое наблюдение членов семьи с гомозиготными мутациями с оценкой функции ЩЖ.
При частичной резистентности к ТТГ в ряде исследований было показано, что наличие повышенного уровня ТТГ может быть достаточным для адекватной выработки гормонов ЩЖ [11][14][35] и не всегда требует назначения заместительной терапии у детей [7].
В 1999 г. создана база данных [36], объединяющая сведения о пациентах с мутациями гена TSHR, включающая клиническую картину и функциональную характеристику мутаций. Последнее обновление базы произведено в 2018 г., зарегистрировано 130 семейных и 60 спорадических случаев нечувствительности к ТТГ с инактивирующими мутациями гена TSHR [37].
Частота мутаций гена TSHR у пациентов с ВГ, обусловленным дисгенезией ЩЖ, по нашим данным, составила 5,3% и во всех случаях сопровождалась гипоплазией ЩЖ. Выявлены 2 ранее не описанные мутации — p.D487N и p.A485D и 2 описанные — p.R450H и p.S49Afs*9, участие которых можно предположить в развитии заболевания. Для подтверждения их патогенности необходимы дополнительные функциональные исследования. В работе изучены пути наследования, наиболее вероятен аутосомно-рецессивный тип наследования.
Учитывая полученные данные и результаты других исследований, в случае полной резистентности к ТТГ назначение левотироксина натрия обязательно. В случае мутации гена TSHR с частичной резистентностью к ТТГ (субклинический гипотиреоз) назначение терапии остается дискутабельным, поскольку в большинстве случаев повышенных уровней ТТГ бывает достаточно для поддержания нормальной концентрации тиреоидных гормонов. Терапия калия йодидом в данном случае неэффективна.
Таким образом, определение генетической основы у детей с незначительным повышением ТТГ по данным скрининга и гипоплазией ЩЖ, а также у людей с неаутоиммунным субклиническим гипотиреозом может помочь в постановке диагноза и определить тактику наблюдения.
Источники финансирования. Исследование выполнено в рамках Государственного задания в части реализации протокола клинической апробации: «Метод гибридной анатомо-функциональной визуализации тиреоидной ткани для топической и функциональной диагностики ВГ у детей» (№2019-15-21), а также при поддержке Благотворительного фонда «Культура благотворительности» в рамках программы «Альфа-Эндо» (молекулярно-генетический блок обследования).
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи.
Участие авторов. Шредер Е.В. — концепция и дизайн исследования, предоставление материалов исследования и анализ данных, интерпретация результатов, написание статьи; Безлепкина О.Б. — концепция и дизайн исследования, редактирование текста, внесение ценных замечаний; Вадина Т.А. — концепция и дизайн исследования, редактирование текста; Конюхова М.Б., Сергеева Н.В. — предоставление материалов исследования; Дегтярев М.В., Солодовникова Е.В., Захарова В.В. — предоставление материалов исследования, редактирование текста, внесение ценных замечаний.
Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
Благодарности. Авторы выносят благодарность Благотворительному фонду «Культура благотворительности» в рамках программы «Альфа-Эндо» за финансирование молекулярно-генетического исследования.
1. Deladoëy J, Bélanger N, Van Vliet G. Random variability in congenital hypothyroidism from thyroid dysgenesis over 16 years in Québec. J Clin Endocrinol Metab. 2007;92(8):3158-3161. doi: https://doi.org/10.1210/jc.2007-0527
2. Clerc J. Imaging the thyroid in children. Best Pract Res Clin Endocrinol Metab. 2014;28(2):203-220. doi: https://doi.org/10.1016/j.beem.2013.04.011
3. Barry Y, Bonaldi C, Goulet V, et al. Increased incidence of congenital hypothyroidism in France from 1982 to 2012: a nationwide multicenter analysis. Ann Epidemiol. 2016;26(2):100-105.e4. doi: https://doi.org/10.1016/j.annepidem.2015.11.005
4. Shreder EV, Vadina TA, Konyukhova MB, et al. Ectopic thyroid gland: clinical features and diagnostics in children. Problems of Endocrinology. 2022;68(3):76-85. (In Russ.). doi: https://doi.org/10.14341/probl12876
5. Shreder EV, Vadina TA, Koniukhova MB, et al. Vrozhdennyi gipotireoz — osobennosti vizualizatsii tkani shchitovidnoi zhelezy. In: Sbornik tezisov konferentsii po orfannym zabolevaniiam i detskim endokrinnym zabolevaniiam s mezhdunarodnym uchastiem: «Dostizheniia nauki v praktiku detskogo endokrinologa». 4–5.12.2021. 71 р. (In Russ.).
6. van Trotsenburg P, Stoupa A, Léger J, et al. Congenital hypothyroidism: A 2020–2021 consensus guidelines update—An ENDO-European Reference Network Initiative Endorsed by the European Society for Pediatric Endocrinology and the European Society for Endocrinology. Thyroid. 2021;31(3):387-419. doi: https://doi.org/10.1089/thy.2020.0333
7. Grasberger H, Refetoff S. Resistance to thyrotropin. Best Pract Res Clin Endocrinol Metab. 2017;31(2):183-194. doi: https://doi.org/10.1016/j.beem.2017.03.004
8. Alberti L, Proverbio MC, Costagliola S, et al. Germline mutations of TSH receptor gene as cause of nonautoimmune subclinical hypothyroidism. J Clin Endocrinol Metab. 2002;87(6):2549-2555. doi: https://doi.org/10.1210/jcem.87.6.8536
9. Rapa A, Monzani A, Moia S, et al. Subclinical hypothyroidism in children and adolescents: A wide range of clinical, biochemical, and genetic factors involved. J Clin Endocrinol Metab. 2009;94(7):2414-2420. doi: https://doi.org/10.1210/jc.2009-0375
10. Nicoletti A, Bal M, De Marco G, et al. Thyrotropin-stimulating hormone receptor gene analysis in pediatric patients with non-autoimmune subclinical hypothyroidism. J Clin Endocrinol Metab. 2009;94(11):4187-4194. doi: https://doi.org/10.1210/jc.2009-0618
11. Tenenbaum-Rakover Y, Grasberger H, Mamanasiri S, et al. Loss-of-function mutations in the thyrotropin receptor gene as a major determinant of hyperthyrotropinemia in a consanguineous community. J Clin Endocrinol Metab. 2009;94(5):1706-1712. doi: https://doi.org/10.1210/jc.2008-1938
12. Calebiro D, Gelmini G, Cordella D, et al. Frequent TSH receptor genetic alterations with variable signaling impairment in a large series of children with nonautoimmune isolated hyperthyrotropinemia. J Clin Endocrinol Metab. 2012;97(1):E156-E160. doi: https://doi.org/10.1210/jc.2011-1938
13. Da D-Z, Wang Y, Wang M, et al. Congenital Hypothyroidism Patients With Thyroid Hormone Receptor Variants Are Not Rare: A Systematic Review. Inq J Heal Care Organ Provision, Financ. 2021;58(5):004695802110679. doi: https://doi.org/10.1177/00469580211067943
14. Osipovskaya MA, Kiyaev AV, Makretskaya NA, et al. Resistance to thyrotropin: familial case report. Clinical and experimental thyroidology. 2015;11(4):36-39. (In Russ.). doi: https://doi.org/10.14341/ket2015436-39
15. Makretskaya NA, Bezlepkina OB, Kolodkina AA, et al. Study of molecular basis of thyroid dysgenesis. Clinical and experimental thyroidology. 2018;14(2):64-71. (In Russ.). doi: https://doi.org/10.14341/ket9556
16. Vadina TA, Shreder EV, Tiul’pakov MA, et al. Dva sluchaia vrozhdennogo gipotireoza, obuslovlennogo raznymi mutatsiiami gena retseptora TTG. In: Sbornik tezisov konferentsii po orfannym zabolevaniiam i detskim endokrinnym zabolevaniiam s mezhdunarodnym uchastiem: «Dostizheniia nauki v praktiku detskogo endokrinologa». 4–5.12.2021, onlain format. P. 13. (In Russ.).
17. Worth C, Hird B, Tetlow L, et al. G557 Value of scintigraphy in identifying cause of congenital hypothyroidism. In: British society for paediatric endocrinology and diabetes. BMJ Publishing Group Ltd and Royal College of Paediatrics and Child Health; 2019. P. A225.2-A226. doi: https://doi.org/10.1136/archdischild-2019-rcpch.540
18. Shreder EV, Vadina TA, Shiriaeva TIu, et al. Topicheskaia diagnostika tireoidnoi tkani u detei s disgeneziei shchitovidnoi zhelezy pri vrozhdennom gipotireoze. In: Sbornik tezisov XVII Rossiiskoi nauchno-prakticheskoi konferentsii detskikh endokrinologov «Dostizheniia nauki v praktiku detskogo endokrinologa», 12–13.06.2021. Moscow: OOO «Tipografiia pechatnykh del master»; 2021. P. 183. (In Russ.).
19. Narumi S, Muroya K, Abe Y, et al. TSHR mutations as a cause of congenital hypothyroidism in Japan: a population-based genetic epidemiology study. J Clin Endocrinol Metab. 2009;94(4):1317-1323. doi: https://doi.org/10.1210/jc.2008-1767
20. Fang Y, Sun F, Zhang R-J, et al. Mutation screening of the TSHR gene in 220 Chinese patients with congenital hypothyroidism. Clin Chim Acta. 2019;497:147-152. doi: https://doi.org/10.1016/j.cca.2019.07.031
21. Ma SG, Fang PH, Hong B, Yu WN. The R450H mutation and D727E polymorphism of the thyrotropin receptor gene in a Chinese child with congenital hypothyroidism. J Pediatr Endocrinol Metab. 2010;23(12):1339-1344. doi: https://doi.org/10.1515/jpem.2010.209
22. Qiu YL, Ma SG, Liu H, Yue HN. Two novel TSHR gene mutations (p.R528C and c.392+4del4) associated with congenital hypothyroidism. Endocr Res. 2016;41(3):180-184. doi: https://doi.org/10.3109/07435800.2015.1124438
23. Park KJ, Park HK, Kim YJ, et al. DUOX2 mutations are frequently associated with congenital hypothyroidism in the Korean population. Ann Lab Med. 2016;36(2):145-153. doi: https://doi.org/10.3343/alm.2016.36.2.145
24. Parmentier M, Libert F, Maenhaut C, et al. Molecular cloning of the thyrotropin receptor. Science. 1989;246(4937):1620-1622. doi: https://doi.org/10.1126/science.2556796
25. Stein SA, Oates EL, Hall CR, et al. Identification of a point mutation in the thyrotropin receptor of the hyt/hyt hypothyroid mouse. Mol Endocrinol. 1994;8(2):129-138. doi: https://doi.org/10.1210/mend.8.2.8170469
26. Akamizu T, Ikuyama S, Saji M, et al. Cloning, chromosomal assignment, and regulation of the rat thyrotropin receptor: expression of the gene is regulated by thyrotropin, agents that increase cAMP levels, and thyroid autoantibodies. Proc Natl Acad Sci U S A. 1990;87(15):5677-5681. doi: https://doi.org/10.1073/pnas.87.15.5677
27. Rousseau-Merck MF, Misrahi M, Loosfelt H, et al. Assignment of the human thyroid stimulating hormone receptor (TSHR) gene to chromosome 14q31. Genomics. 1990;8(2):233-236. doi: https://doi.org/10.1016/0888-7543(90)90276-z
28. Libert F, Passage E, Lefort A, et al. Localization of human thyrotropin receptor gene to chromosome region 14q3 by in situ hybridization. Cytogenet Cell Genet. 1990;54(1-2):82-83. doi: https://doi.org/10.1159/000132964
29. Rapoport B, Chazenbalk GD, Jaume JC, McLachlan SM. The Thyrotropin (TSH)-Releasing Hormone Receptor: Interaction with TSH and Autoantibodies. Endocr Rev. 1998;19(6):673-716. doi: https://doi.org/10.1210/edrv.19.6.0352
30. Abramowicz MJ, Duprez L, Parma J, et al. Familial congenital hypothyroidism due to inactivating mutation of the thyrotropin receptor causing profound hypoplasia of the thyroid gland. J Clin Invest. 1997;99(12):3018-3024. doi: https://doi.org/10.1172/JCI119497
31. Lábadi Á, Grassi ES, Gellén B, et al. Loss-of-function variants in a Hungarian cohort reveal structural insights on TSH Receptor Maturation and Signaling. J Clin Endocrinol Metab. 2015;100(7):E1039-E1045. doi: https://doi.org/10.1210/jc.2014-4511
32. Chang WC, Liao CY, Chen WC, et al. R450H TSH receptor mutation in congenital hypothyroidism in Taiwanese children. Clin Chim Acta. 2012;413(11-12):1004-1007. doi: https://doi.org/10.1016/j.cca.2012.02.027
33. Tsunekawa K, Yanagawa Y, Aoki T, et al. Frequency and clinical implication of the R450H mutation in the thyrotropin receptor gene in the Japanese population detected by Smart Amplification Process 2. Biomed Res Int. 2014;2014:964635. doi: https://doi.org/10.1155/2014/964635
34. Makretskaya N, Bezlepkina O, Kolodkina A, et al. High frequency of mutations in ‘dyshormonogenesis genes’ in severe congenital hypothyroidism. PLoS One. 2018;13(9):e0204323. doi: https://doi.org/10.1371/journal.pone.0204323
35. Mizuno H, Kanda K, Sugiyama Y, et al. Longitudinal Evaluation of Patients with a Homozygous R450H Mutation of the TSH Receptor Gene. Horm Res Paediatr. 2009;71(6):318-323. doi: https://doi.org/10.1159/000223415
36. Thyroid stimulating hormone receptor mutation database, 2023 [cited 01/12/2022]. Available from: tsh-receptor-mutation-database.org.
37. Stephenson A, Lau L, Eszlinger M, Paschke R. The thyrotropin receptor mutation database update. Thyroid. 2020;30(6):931-935. doi: https://doi.org/10.1089/thy.2019.0807
Шредер Екатерина Владимировна.
117036, Москва, ул. Дм. Ульянова, д. 11
Нет
Вадина Татьяна Алексеевна – кандидат медицинских наук.
Москва
Нет
Солодовникова Екатерина Николаевна.
Москва
Нет
Захарова Виктория Витальевна - кандидат медицинских наук.
Москва
Нет
Дегтярев Михаил Владимирович.
Москва
Нет
Конюхова Марина Борисовна - кандидат медицинских наук.
Москва
Нет
Сергеева Наталия Вячеславовна.
Москва
Нет
Безлепкина Ольга Борисовна – доктор медицинских наук, профессор.
Москва
Нет
|
|
1. Рисунок 1. Структура дисгенезии щитовидной железы у детей с врожденным гипотиреозом. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(90KB)
|
Метаданные ▾ | |
|
|
2. Рисунок 2. Родословные пациентов с мутацией в гене TSHR (NP_000360.2). | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(202KB)
|
Метаданные ▾ | |
|
|
3. Рисунок 3. Модель TSHR с расположением подтвержденных и предполагаемых инактивирующих мутаций, выявленных у пациентов с резистентностью к ТТГ (адаптирован из [7]; с изменениями). | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(559KB)
|
Метаданные ▾ | |
Шрёдер Е.В., Вадина Т.А., Солодовникова Е.Н., Захарова В.В., Дегтярев М.В., Конюхова М.Б., Сергеева Н.В., Безлепкина О.Б. Патогенные варианты гена TSHR у детей с дисгенезией щитовидной железы. Проблемы Эндокринологии. 2023;69(1):76-85. https://doi.org/10.14341/probl13210
Shreder E.V., Vadina T.A., Solodovnikova E.N., Zakharova V.V., Degtyarev M.V., Konyukhova M.B., Sergeeva N.V., Bezlepkina O.B. Pathogenic TSHR variants in children with thyroid dysgenesis. Problems of Endocrinology. 2023;69(1):76-85. (In Russ.) https://doi.org/10.14341/probl13210

 |
![]()
![]()

![]()
117292, Российская Федерация, Москва, ул. Дм. Ульянова, д.11



































