




Содержание
Перейти к:
https://doi.org/10.14341/probl13273
Перейти к:
За последние годы проводится большое количество исследований по изучению молекулярно-генетических аномалий в АКТГ-секретирующих опухолях гипофиза. В данном обзоре представлен комплексный анализ результатов исследований полного экзома (герминальные и соматические мутации, хромосомные аномалии в кортикотропиномах, развившихся в составе наследственных синдромов МЭН 1, 2, 4, DICER1, комплекс Карни и пр., и изолированных опухолях соответственно) и транскриптома (специфичные профили экспрессии генов гормонально активных и неактивных кортикотропином, регуляция клеточных циклов и сигнальных путей). Современные технологии (секвенирование следующего поколения — NGS) позволяют изучить состояние микроРНКома, ДНК метилома. Таким образом, показан широкий спектр фундаментальных исследований, результаты которых позволяют определить и осмыслить ключевые, ранее известные и новые механизмы патогенеза и биомаркеры кортикотропином. Дана характеристика наиболее перспективным молекулярно-генетическим факторам, которые возможно использовать в клинической практике для скрининга и более ранней диагностики наследственных синдромов и изолированных кортикотропином, дифференциальной диагностики различных форм эндогенного гиперкортицизма, чувствительности к уже существующим и потенциальным видам терапии и персонализированного определения исхода болезни Иценко-Кушинга.
Лапшина А.М. Молекулярно-генетические аномалии в кортикотропных опухолях гипофиза: фундаментальные исследования и перспективы использования в клинической практике. Проблемы Эндокринологии. 2024;70(3):23-30. https://doi.org/10.14341/probl13273
Lapshina A.M. Molecular genetic abnormalities in ACTH-secreting pituitary tumors (corticotropinomas): fundamental research and prospects for use in clinical practice. Problems of Endocrinology. 2024;70(3):23-30. (In Russ.) https://doi.org/10.14341/probl13273
За последние годы был достигнут значительный прогресс в понимании молекулярно-биологических аномалий в кортикотропиномах и других опухолях гипофиза. К ним относят генетические и эпигенетические факторы, драйверные соматические мутации, вариабельность количества нуклеотидов в составе хромосом, метилирование генов, регуляции микроРНК и факторов транскрипции.
Болезнь Иценко-Кушига — это тяжелое многосимптомное заболевание гипоталамо-гипофизарного происхождения, вызванное АКТГ-секретирующей опухолью гипофиза (кортикотропиномой), которая стимулирует избыточную секрецию гормонов коры надпочечников. Среди всех опухолей гипофиза кортикотропиномы составляют 10–15%. Методом выбора лечения является нейрохирургическое удаление опухоли. Кортикотропиномы — это одна из наиболее часто встречаемых опухолей гипофиза в операционном материале. По данным отдела фундаментальной патоморфологии ГНЦ НМИЦ эндокринологии, на долю АКТГ-секретирующих опухолей гипофиза приходится до 39,4% случаев у взрослых и 62,5% у детей. Это позволяет использовать как образцы нефиксированной формалином ткани опухоли, так и из парафиновых блоков в совокупности с исследованием крови для выполнения молекулярно-генетического тестирования.
Использование знаний в отношении генетических аномалий у пациентов с кортикотропиномами начинает входить в клиническую практику. На сегодняшний день наиболее изучены герминальные мутации в опухолях гипофиза, развивающиеся как наследственные заболевания в составе синдромов (различные варианты синдрома множественных эндокринных неоплазий (МЭН), комплекс Карни, синдром МакКьюна-Олбрайта, 3Р ассоциации: феохромоцитома\параганглиома\опухоль гипофиза, DICER1, семейные изолированные опухоли гипофиза). Однако группа этих опухолей достаточно малочисленна и составляет всего 5% от всех новообразований аденогипофиза. Большая часть представлена спорадическими опухолями с наличием соматических мутаций в различных генах и менее изучена по сравнению с аденомами гипофиза, развившихся в составе указанных синдромов.
Рецензируемые исследовательские статьи были взяты из Национального центра биотехнологий «Информационная база данных» (https://pubmed.ncbi.nlm.nih.gov/, за 2018-2022 гг). Основная цель анализа литературы — всесторонний поиск молекулярно-генетических маркеров в отношении гормонально активных и гормонально неактивных кортикотропином из исследований, проведенных в предыдущие годы, поэтому для статей не было установлено ограничений по дате. Ключевые слова для статей по геномике включали: «генетика кортикотропином/аденом гипофиза/нейроэндокринных опухолей гипофиза, семейных кортикотропином/аденом гипофиза/нейроэндокринных опухолей гипофиза», «секвенирование кортикотропином/аденом гипофиза/нейроэндокринных опухолей гипофиза». Для статей по транскриптомике ключевыми словами являлись «транскриптом кортикотропином/аденом гипофиза/нейроэндокринных опухолей гипофиза»», а также «секвенирование РНК кортикотропином/аденом гипофиза/нейроэндокринных опухолей гипофиза». Для классификации различных подтипов нейроэндокринных опухолей гипофиза и выделения среди них вариантов кортикотропином в обзоре использовалась классификация опухолей эндокринных органов Всемирной организации здравоохранения (ВОЗ) от 2022 г.
Цель данной работы — представить результаты комплексного анализа полного экзома и транскриптома гормонально активных и неактивных кортикотропином, а также данные современных технологий, посвященные изучению микроРНКома, ДНК метилома. В то же время в нашей работе мы попытались проследить, каким образом фундаментальные исследования позволяют определить ключевые механизмы патогенеза и биомаркеры кортикотропином, и как возможно использовать эти показатели в клинической практике для скрининга и более ранней диагностики наследственных синдромов и изолированных кортикотропином, дифференциальной диагностики различных форм эндогенного гиперкортицизма, чувствительности к уже существующим и потенциальным видам терапии, определении персонализированного исхода гормонально активных и неактивных кортикотропином.
Среди наследственных синдромов для кортикотропином описаны синдромы МЭН1, 2А, 2Б и 4, DICER1, а также синдром семейных изолированных опухолей гипофиза (FIPA), комплекс Карни и синдром Линча.
Синдром множественных эндокринных неоплазий 1 типа (МЭН1) наследуется по аутосомно-доминантному типу с распространенностью около 1:10 000–1:100 000 населения. Опухоли гипофиза развиваются у 30–40% таких пациентов, тогда как гиперплазия или опухоли околощитовидных желез и нейроэндокринные опухоли желудочно-кишечного тракта возникают гораздо чаще (до 90 и 60% соответственно). Среди опухолей гипофиза в составе синдрома МЭН1 кортикотропиномы развиваются достаточно редко. По данным Vergès B и соавт., их доля составляет около 4%. Наиболее часто диагностируют пролактиномы (40–60%), далее следуют гормонально неактивные опухоли гипофиза (15–40%) и соматотропиномы (5–10%). Клинически такие опухоли характеризуются крупными размерами, чаще поражают молодых пациентов, могут быть множественными или способны секретировать 2 и более гормонов аденогипофиза [1].
У 90% пациентов с МЭН1 синдромом определяется герминальная гетерозиготная мутация в гене MEN1, который расположен на длинном плече 11 хромосомы (11q13) и является геном-супрессором. MEN1 кодирует синтез белка менина, который располагается в ядре клетки и участвует в регуляции клеточной транскрипции, делении и пролиферации клеток, поддержании геномной стабильности. Последние годы появились коммерческие антитела к менину для выполнения иммуногистохимического исследования в ткани опухоли с целью скрининга данной мутации [2].
В литературе описаны единичные случаи кортикотропином (1 у взрослого и 1 у ребенка), развившихся в рамках синдромов МЭН 2А и 2В. Данные синдромы связаны с мутацией онкогена RET. Он кодирует трансмембранный рецептор, связанный с активностью тирозинкиназы, которая действует как протоонкоген [3][4].
Синдром МЭН4 — это редкое генетическое заболевание, которое развивается у 1,5–3% пациентов при отсутствии мутаций в гене MEN1. Патология вызвана мутацией в гене CDKN1B, который расположен в хромосоме 12q13 и кодирует циклин-зависимую киназу р27. Этот фермент регулирует клеточный цикл от G1 до S фазы митоза. В литературе описаны единичные случаи таких пациентов с АКТГ-секретирующими опухолями [5].
DICER1 синдром — это редкое, аутосомно доминантное заболевание, обусловленное герминальной гетерозиготной мутацией в гене DICER1. Синдром включает в себя различные доброкачественные и злокачественные опухоли: плеврально-легочная бластома, опухоли яичников из клеток стромы полового тяжа (сертоликлеточные и лейдигоклеточные), кистозная нефрома, узловая гиперплазия и злокачественные дифференцированные опухоли щитовидной железы, бластома гипофиза, назальная хондромезенхимальная гамартома, медуллоэпителиома реснитчатого тела, почечная саркома, эмбриональная рабдомиосаркома и пинеалобластома. Бластома гипофиза впервые описана в 2008 г. [6], гистологически представлена Ратке-подобными эпителиальными клетками, формирующими розетки или железисто-подобные структуры; среди них имеются АКТГ-позитивные клетки (реже — клетки с экспрессией гормона роста). В составе DICER1 синдрома бластомы гипофиза диагностируют редко (менее 1%). Они характеризуются крупными размерами и тяжелым течением БИК у детей с медианой возраста 8 месяцев (в диапазоне от 7 до 24 месяцев), смертность достигает 40%. Мутация в гене DICER1 является патогномоничной для данного синдрома. Ген расположен на длинном плече 14 хромосомы (14q32.13), кодирует цитоплазматическую эндорибонуклеазу, которая отвечает за созревание и модуляцию экспрессии микроРНК на посттранскрипционном уровне. Недавно в литературе описан DICER1 синдром у молодого взрослого пациента, обусловленный герминальной гетерозиготной мутацией в гене DICER1 [7].
Герминальная мутация в гене AIP, который кодирует белок, взаимодействующий с арилуглеводородным рецептором, может быть причиной возникновения опухолей гипофиза в 15–30% семей с синдромом семейных изолированных аденом гипофиза. Мутация в данном гене чаще описана у пациентов в пораженных семьях с акрогигантизмом, реже — с пролактиномами или в опухолях со смешанной секрецией гормона роста и пролактина. Для этих новообразований характерны крупные размеры, они резистентные к медикаментозной терапии, поражают молодых людей. Крайне редко такие мутации возникают у пациентов с БИК: в литературе описаны единичные случаи у детей и взрослых. В исследование L. Cazabat [8] были включены 443 пациента, у которых определяли наличие мутаций в гене AIP. Среди них выявили 44 пациента с БИК (10%), у троих из которых обнаружили данную мутацию. Эти пациенты были моложе 40 лет (на момент манифестации заболевания возраст составил 13, 21 и 39 лет). Среди пациентов с кортикотропиномами преобладали те, у которых фиксировались микроаденомы (2 микро- и 1 макроаденома), в отличие от больных с другими гистологическими типами макроаденом гипофиза. Были обнаружены нуклеотидные замены по типу миссенс-мутаций (n=2) и сдвигу рамки считывания (n=1) в гене AIP.
Исследованы и описаны несколько механизмов, приводящих к резистентности к аналогам соматостатина у таких пациентов. К ним относят снижение экспрессии ингибирующего G-протеина, фосфодиэстеразы, а также нарушения сигнального пути протеинкиназы А и ZAC1.
Существуют еще 2 наследственных синдрома, в рамках которых отмечают пациентов с кортикотропиномами. Это комплекс Карни и синдром Линча. Комплекс Карни — это наследственный синдром, обусловленный инактивирующими мутациями в гене PRKAR1A, который кодирует альфа-регуляторную субъединицу цАМФ-зависимой протеинкиназы А. В составе комплекса Карни, кроме шванном, миксом и пигментации кожи, диагностируют кортикостеромы. За последние годы также появились упоминания о единичных кортикотропиномах [9].
Синдром Линча развивается в результате герминальных мутаций в так называемых генах репарации ДНК (MSH2, MLH1, MSH6, PMS2 и EPCAM) и ассоциируется с наследственной предрасположенностью к развитию карцином различной локализации. В литературе описаны случаи пациентов с синдромом Линча с наличием инвазивных макрокортикотропином или кортикотрофной карциномой, у которых обнаружили герминальные мутации в генах MLH1 и MSH2 [10][11].
Таким образом, пациенты с кортикотропиномами молодого (менее 30 лет) и детского возраста могут являться кандидатами для молекулярного тестирования с целью идентификации наследственных синдромов. В качестве скрининга возможно проводить иммуногистохимическое исследование на образцах ткани опухоли с антителами к менину (синдром МЭН1), р27 (синдром МЭН4), MSH2, MLH1, MSH6, PMS2 (синдром Линча).
Наиболее изученными и распространенными среди спорадических АКТГ-секретирующих опухолей гипофиза являются соматические мутации в генах убиквитин-специфической протеазы 8 (USP8), убиквитин-специфической протеазы 48 (USP 48), BRAFV600E, ATRX.
Полное секвенирование экзома выявило, что 20–60% кортикотропином являются носителями соматической мутации в гене USP8. Данная мутация является специфичной для кортикотропином и не обнаружена в других типах опухолей аденогипофиза или АКТГ-эктопических опухолей негипофизарной локализации.
«Горячая точка» гена USP8 расположена в 14 экзоне связывающего домена 14-3-3. В белке USP8 дикого типа в домене 14-3-3 происходят конформационные изменения, которые позволяют USP8 блокировать его собственную каталитическую активность. Потеря связывающего участка 14-3-3 при наличии мутации в гене USP8 повышает их деубиквитиназную активность и обеспечивает доступ к протеазам, которые расщепляют его до С-концевого фрагмента длиной 40 кДА с высокой каталитической способностью. Этот процесс нарушает деградацию EGFR (рецептор эпидермального фактора роста), стимулирует транскрипцию проопиомеланокортина (ПОМК) и синтеза АКТГ в опухоли. Надо отметить, что выраженная стимуляция митотической активности и, как следствие, высокая пролиферативная активность в опухолевых кортикотрофных клетках не обнаружена [12].
Кортикотропиномы, содержащие мутации в гене USP8, чаще возникают у женщин, меньше по размеру и неинвазивные. Уровни секреции АКТГ до операции выше по сравнению с показателями пациентов, у которых кортикотропиномы не обладали мутацией в гене USP8. С другой стороны, случаи с наличием соматических мутаций в гене USP8 сопровождаются более высокими уровнями кортизола после удаления опухоли и как следствие рецидивом или продолженным ростом новообразования. Эти данные подтверждаются наличием указанной мутации у 50% пациентов с продолженным ростом кортикотропином при синдроме Нельсона. В то же время показано, что опухоли без мутаций в гене USP8 обладают более крупными размерами с инвазивным ростом в сфеноидальный синус, ремиссия развивается реже [13].
Что касается детей с наличием БИК, то, по данным Faucz FR [14], кортикотропиномы, несущие соматические мутации в гене USP8, обнаружены у 13 из 45 пациентов (28%). Однако в литературе описан клинический случай кортикотропиномы у 16-летней девушки с наличием гетерозиготной герминальной мутации в гене USP8 [15].
Кортикотропиномы, содержащие мутации в гене USP8, обладают высоким уровнем экспрессии рецепторов соматостатина 5 подтипа и О6-метилгуанин ДНК метилтрансферазы (MGMT), следовательно, чувствительны к терапии пазиреотидом (лиганд к рецепторам соматостатина 5 подтипа) и темозоламидом соответственно.
Мутация в гене USP8 может являться мишенью для лечения с помощью низкомолекулярных ингибиторов, демонстрирующих антипролиферативную и антисекреторную эффективность in vitro. В качестве ингибитора EGFR может использоваться гефитиниб для терапии пациентов с кортикотропиномами, содержащими мутации в гене USP8. Показано прямое ингибирование EGFR в первичных культурах клеток кортикотропином, содержащих мутацию в указанном гене, с последующим ослаблением высвобождения АКТГ [16][17].
Среди кортикотропином с помощью секвенирования следующего поколения (NGS) была выявлена вторая «горячая точка» в гене, кодирующем другую деубиквитиназу, — USP48. Мутация в гене USP48 наблюдаются в 4–23% случаев кортикотропином (без мутаций в гене USP8) и является миссенс-мутацией, включающей M415I/V субституцию. Мутантный ген усиливает транскрипционную активность гена POMC, предшественника АКТГ, за счет митоген-активируемой протеинкиназы (MAPK). Для кортикотропином, несущих мутацию в гене USP48, характерны меньшие размеры и более высокая чувствительность к стимуляции кортикотропин-рилизинг гормона (КРГ-физиологического стимулятора синтеза и секреции АКТГ в кортикотрофных клетках). In vitro на модели клеток кортикотропином (MET415lle) с наличием мутации в гене USP48 определялось умеренное повышение секреции АКТГ. На фоне стимуляции КРГ уровни ПОМК и АКТГ были значительно выше базальных значений, секретируемые клетками кортикотропином с наличием мутации в гене USP48, по сравнению с клетками без мутаций в данном гене (р<0,01). На основании этих данных можно предположить, что мутация в гене USP48 может индуцировать онкогенез кортикотропином за счет высокой чувствительности к стимулу КРГ со стороны гипоталамуса. В экспериментах также было показано значение Gli1 (глиома-ассоциированный онкоген), медиатора сигнального пути Sonic Hedgehog (SHH). Gli1 считается ключевым фактором, который влияет на процесс убиквитинирования и потенцирует синтез АКТГ за счет активации сигнального пути SHH в кортикотропиномах, содержащих мутации в гене USP48. Предполагается подобное действие Gli1/SHH сигнального пути и в кортикотропиномах с наличием мутации в гене USP8 [18][19].
Около 16% случаев кортикотропином содержат мутацию в гене BRAF V600E при отсутствии мутаций в генах USP8 и USP48. Данная мутация усиливает действие протоонкогена с тирозин-киназной активностью, что запускает MAPK-сигнальный путь и активирует транскрипцию гена POMC и синтез АКТГ.
На сегодняшний день существует проблема определения прогноза течения БИК. Анализ совокупности данных об особенностях клинического течения, МРТ (размер опухоли и/или инвазивный рост), гистологические варианты кортикотропином (плотно- и редкогранулированные опухоли, из Круковских клеток), показатели пролиферативной активности (количество митозов и индекс метки Ki-67, экспрессия Р53) могут не соответствовать характеру течения заболевания (ремиссия, персистирующий гиперкортицизм или рецидив заболевания). В исследовании Uzilov V [20] изучали наличие мутации в генах USP8 и TP53 в кортикотропиномах, в том числе и в когорте опухолей с агрессивным течением. В результате было показано, что одновременное содержание указанных мутаций в одной и той же кортикотропиноме исключалось. Это подтверждает независимую работу данных драйверных генов. Многочисленные исследования доказывают наличие мутации в гене ТР 53 во многих карциномах различной локализации. Среди АКТГ-секретирующих новообразований гипофиза обнаружены случаи с инактивирующей соматической драйверной мутацией в гене ТР53. Группа этих опухолей (n=5 из 22) была представлена инвазивными макроаденомами с персистирующим или рецидивирующим течением заболевания. Опухоли были позитивны к P53 (при иммуногистохимическом исследовании) и обладали высоким пролиферативным индексом Ki-67 (более 3%). В 3 случаях диагностировали синдром Нельсона, у 1 пациента выявлена карцинома. На основании этих данных авторы определили взаимосвязь между наличием мутации в гене ТР53 и агрессивным течением БИК, также предположили, что позитивная иммуноэкспрессия Р53 ассоциирована с инвазивным и агрессивным характером роста опухолей. Однако необходимо дальнейшее изучение взаимосвязи между наличием мутации в гене ТР53, позитивной иммуноэкспрессией Р53 в клетках опухоли и агрессивным течением БИК.
Данное исследование также продемонстрировало, что кортикотропиномы, несущие соматические мутации в гене USP8, были более генетически стабильны по сравнению с опухолями без мутаций в этом гене и обладали благоприятным течением БИК. Часть опухолей (22,7%) содержали мутацию в гене TP53 и обладали высоким уровнем хромосомной нестабильности. В этих случаях наблюдалось персистирующее или рецидивирующее течение заболевания.
В ряде работ показана связь с наличием мутации в гене ATRX (alpha thalassemia/mental retardation syndrome X-linked — синдром альфа-таласемии и умственной отсталости X-сцепленного типа), регулирующий ремоделирование хроматина и поддержание структуры и функции теломеры. Инактивация гена ATRX в опухолях приводит к дестабилизации теломеры и стимулирует процесс альтернативного удлинения теломер, что запускает состояние бессмертия опухолевых клеток. Соматические мутации в гене ATRX ассоциируются с астроцитомами у взрослых и такими нейроэндокринными опухолями (НЭО), как НЭО поджелудочной железы, феохромоцитомы/параганглиомы, нейробластомы. Предполагается, что аномалии в структуре гена ATRX являются предикторами агрессивного поведения указанных новообразований [21][22][23][24][25].
В работах Casar-Borota O. и соавт. [26][27], посвященных изучению иммуноэкспрессии белка ATRX при иммуногистохимическом исследовании и мутации гена ATRX (при NGS) в различных опухолях гипофиза, показано выпадение иммуноэкспрессии этого белка именно в опухолях с агрессивным течением (13%) или в карциномах (28%). Причем большинство были представлены кортикотропными опухолями (32%) по сравнению с Pit-1-позитивными новообразованиями (8%) (соматотропиномы, пролактиномы, тиреотропиномы). Отсутствие экспрессии ATRX было подтверждено наличием молекулярных дефектов гена ATRX по типу потери функции (нонсенс-мутации, вставки со смещением рамки считывания, крупными интрагенными делециями практически всего гена — 22-28 экзонов из 36). В результате авторы предложили использовать белок ATRX как иммуногистохимический маркер — предиктор высокоагрессивных или потенциально метастазирующих кортикотропных опухолей гипофиза.
Таким образом, на сегодняшний день актуальным является изучение особенностей клинического течения, гистологических вариантов и прогноза течения заболевания у пациентов с кортикотропиномами, несущими мутации в генах USP8, USP48, BRAF V600E, ТР 53, ATRX. Изучение молекулярно-генетических аномалий приведет к более глубокому пониманию онкогенеза кортикотропином, выявлению дополнительных свойств опухолей, ассоциированных с неблагоприятным течением БИК. Это будет способствовать активному наблюдению за пациентами — носителями мутаций в генах USP8, USP48 и BRAF и своевременному назначению более агрессивной терапии, а белки-продукты мутаций в указанных генах будут являться мишенями для ингибиторов EGFR и BRAF.
За последнее десятилетие накоплено немало доказательств в отношении нарушения регуляции экспрессии микроРНК (миРНК), участвующей в развитии и прогрессии опухолей гипофиза. МиРНК являются некодирующими РНК молекулами длиной 18-24 нуклеотидов, которые регулируют экспрессию генов на посттранскрипционном уровне и контролируют такие процессы, как дифференцировка, пролиферация и апоптоз в клетке. МиРНК влияют на стабильность и трансляцию матричной РНК (мРНК) при помощи связывания с регуляторными элементами, расположенными в 3`нетранслируемых регионах подконтрольных транскриптов. Эти молекулы существуют как внутри клетки, так и за ее пределами в различных биологических жидкостях (плазма, сыворотка, моча, слюна, семенная жидкость, ликвор и др.). Доступность указанных биологических сред позволяет использовать миРНК, как неинвазивные биомаркеры различных заболеваний, в том числе опухолей. Для каждого вида опухолей гипофиза характерен свой специфический профиль экспрессии миРНК. В ряде исследований показано, что аберрантная экспрессия определенных миРНК ассоциируется с размером опухоли, наличием инвазивного роста и чувствительности к различным видам терапии.
В исследовании Sh. Vetrivel и соавт. [28] изучалась экспрессия миРНК для дифференциальной диагностики БИК и СИК, БИК и АКТГ-эктопического синдрома.
Профили циркулирующих миРНК изучались в образцах сыворотки у пациентов с СИК, БИК и контрольной группы. В результате была идентифицирована миРНК 182-5p в качестве потенциального неинвазивного маркера БИК, которая может использоваться для дифференциальной диагностики различных форм эндогенного гиперкортицизма.
В работе Малыгиной А.А. и соавт. [29] проводился поиск миРНК, специфичных для различных форм АКТГ-зависимого гиперкортицизма (БИК и АКТГ-эктопического синдрома). В результате выявлены возможные кандидаты миРНК (miR-383-3p, miR-1229-3p, miR-1203, miR-639, miR-4290, miR-6717-5p, miR-302c-3p), которые предполагается использовать в качестве инструмента в дополнение к уже известным и широко применяемым методам (МРТ головного мозга и селективный забор крови из нижних каменистых синусов) для дифференциальной диагностики форм АКТГ-зависимого гиперкортицизма [30].
Важно отметить исследование F. Garbicz и соавт. [31], в котором определялась иммуноэкспрессия MCM7 (белок 7 для поддержания мини-хромосом участвует в инициации репликации генома эукариот) и экспрессия кластера миРНК miR-106b~25 (miR-106b-5p, miR-93-5p, miR-93-3p and miR-25-3p) при помощи количественной ПЦР в реальном времени в ткани кортикотропином. МСМ7 является геном-мишенью для кластера миРНК miR-106b~25. В АКТГ-секретирующих опухолях, по данным МРТ, определяли степень инвазивного роста по шкале Knosp. При помощи иммуногистохимического исследования выделяли гистологические варианты кортикотропином (плотногранулированные, редкогранулированные и опухоли из круковских клеток), проводили анализ индекса метки Ki-67 и p53. В результате была выявлена повышенная экспрессия МСМ7 и высокий индекс метки Ki-67 в инвазивных кортикотропиномах и в кортикотропиномах из круковских клеток. Экспрессия miR-93-5p была значительно повышена в инвазивных опухолях по сравнению с неинвазивными. Кроме того, все четыре миРНК из кластера miR-106b~25 продемонстрировали гиперэкспрессию в аденомах из круковских клеток. Примечательно, что MCM7 и miR-106b-5p коррелировали со степенью инвазивного роста, оцененного по шкале Knosp. Комбинация экспрессии MCM7, Ki-67 и кластера miR-106b~25 точно дифференцирует инвазивные опухоли от неинвазивных и обладает значительной дискриминационной способностью прогнозировать послеоперационный рецидив/прогрессирование опухоли.
Таким образом, эпигенетические маркеры обладают специфическими характеристиками, которые позволяют дифференцировать формы эндогенного гиперкортицизма (АКТГ-зависимые и АКТГ-независимые) и являются прогностическими биомаркерами.
Ранее в исследованиях использовался моноомиксный подход, основанный на анализируемых клинических или патоморфологических критериях. Последние годы ученые предлагают применять так называемые мультиомиксные методы исследования, которые включают в себя исследование полного экзома и транскриптома. Все эти исследования выполняются при помощи секвенирования следующего поколения (NGS). Т.е. используются комбинации «омиксных» методов с учетом клинической картины, патоморфологических особенностей, характера течения заболеваний (индолентный или агрессивный), чувствительности к различным видам терапии. Мультиомиксный подход в изучении опухолей гипофиза в целом и кортикотропином в частности предполагает детальное понимание онкогенеза. Анализ полного экзома демонстрирует наличие наследственных или соматических драйверных мутаций, хромосомных аномалий. Транскриптом является частью генома, который транскрибируется на РНК с последующей регуляцией генов и синтеза их продуктов (набор белков). МиРНКом представлен гипер- или гипоэкспрессией миРНК. Транскриптом и миРНКом отражает различные свойства опухолей, например происхождение или способность к прогрессии. ДНКметилом демонстрирует уровень гипер- или гипометилирования различных генов, характерных для определенных типов опухолей гипофиза [32].
В исследовании M. Neou [33] проводился совокупный анализ типа секреции опухолей гипофиза, иммунофенотипа, наличие ремиссии или персистенции заболевания, полного экзома и РНК последовательностей, а также эпигенетического профиля и ДНК метилирования. В данной работе на основании комплексного анализа генома опухолей гипофиза было введено такое понятие, как пангеномная классификация. В результате показано, что кортикотропиномы без мутаций в гене USP8 обладали более агрессивным течением с преобладанием инвазивного роста в сфеноидальный синус по сравнению опухолями, содержащими мутацию в гене USP8. Отличия в отношении инвазивного роста в кавернозный синус и уровня индекса метки Ki-67 в этих группах не обнаружены. Инвазивный рост кортикотропиномы без мутации в гене USP8 ассоциировался со способностью этих опухолей к эпителиально-мезенхимальному транзиту (снижению степени дифференцировки). Среди кортикотропином с наличием мутации в гене USP8 выявлен более высокий уровень экспрессии рецепторов соматостатина 5 подтипа по сравнению с опухолями без мутаций в этом гене, тем самым подтверждая потенциальную значимость USP8 статуса в отношении чувствительности к пазиреотиду. При анализе структуры хромосом нарушения в виде потери или увеличения участков были более выражены в гормонально активных кортикотропиномах по сравнению с «немыми» новообразованиями. В данной работе хромосомные нарушения не ассоциировались ни с иммунофенотипом опухоли, ни с агрессивным течением. Профили миРНК и метилирование ДНК коррелировали с секреторными типами опухолей, и кортикотропиномы обладали своим уникальным набором экспрессии этих молекул. Кортикотропиномы продемонстрировали обратную корреляцию между уровнем метилирования и хромосомными аномалиями. Такие характеристики транскриптома, как низкий уровень оксидативного фосфорилирования, воспаления и эпителиально-мезенхимального транзита, значительно отличались между различными типами кортикотропином в зависимости от USP8 статуса. При изучении транскриптома в клинически гормонально неактивных кортикотропиномах были выявлены черты, характерные как для кортикотропином, так и гонадотропином. Эти свойства были подтверждены наличием коэкспрессии иммуногистохимических маркеров, которые выявляются как в гонадотропиномах, так и в кортикотропиномах (АКТГ, TPIT, GATA3). В исследовании Salomon M.P. и соавт. [34] были выявлены особенности в метиломе кортикотропином: определены участки гипометилирования промоторов гена POMC. В то же время проводилось сравнение уровней экспрессии POMC и метилирования промотора гена POMC среди кортикотропином с мутациями и без в гене USP8. В результате разница по указанным показателям в зависимости от USP8 статуса не обнаружена и сделан вывод о том, что ДНК метилирование промотора POMC и последующая его экспрессия и синтез АКТГ происходят независимо от указанного состояния. Совокупность современных молекулярных методов для изучения кортикотропином с учетом использования типов нуклеиновых кислот или РНК молекул, а также существующие дизайны исследований, представлены на рисунке 1.
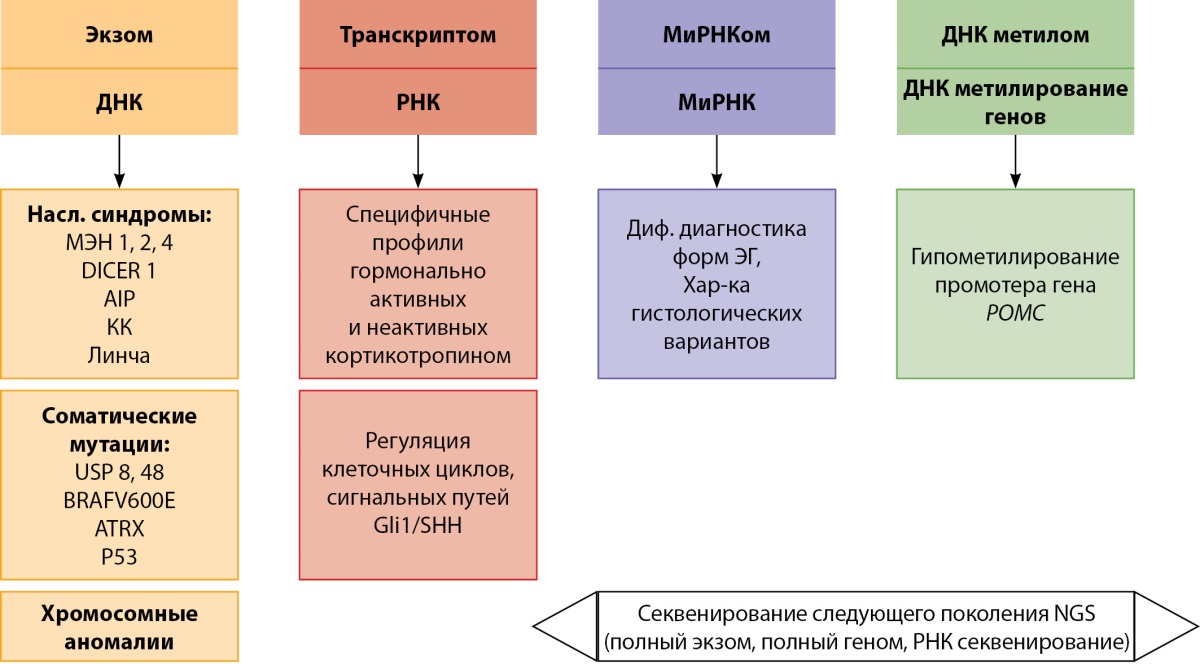
Рисунок 1. Мультиомиксные методы изучения кортикотропином.
Примечание: насл. синдромы — наследственные синдромы, КК — комплекс Карни, диф. диагностика — дифференциальная диагностика, ЭГ — эндогенный гиперкортицизм, хар-ка — характеристика. За основу данного рисунка взято изображение из статьи Peculis R, Niedra H, Rovite V. Large scale molecular studies of pituitary neuroendocrine tumors: novel markers, mechanisms and translational perspectives. Cancers. 2021;13,1395. doi: https://doi.org/10.3390/cancers13061395 [32] и переработано автором.
Таким образом, многочисленные результаты фундаментальных как моно-, так и мультиомиксных исследований кортикотропином представляют комплексный детальный анализ и позволяют определить новые, ранее не описанные молекулярные варианты опухолей, способствуют более глубокому пониманию биологических механизмов. Это потенциально приведет к усовершенствованию диагностики и лечения пациентов с болезнью Иценко-Кушинга в клинической практике.
Источники финансирования. Грант Минобрнауки, соглашение №075-15-2022-310 от 20.04.2022.
Конфликт интересов. Автор декларирует отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Участие авторов. Автор одобрил финальную версию статьи перед публикацией, выразил согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
1. Vergès B, Boureille F, Goudet P, et al. Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. Journal of Clinical Endocrinology and Metabolism. 2002;87(2):457-465. doi: https://doi.org/10.1210/jcem.87.2.8145
2. Asa SL, Mete O, Perry A, Osamura RY. Overview of the 2022 WHO Classification of Pituitary Tumors. Endocrine Pathology. 2022;33(1):6-26. doi: https://doi.org/10.1007/s12022-022-09703-7
3. Naziat A, Karavitaki N, Thakker R, et al. Confusing genes: a patient with MEN2A and Cushing’s disease. Clinical Endocrinology. 2013;78(6):966-968. doi: https://doi.org/10.1111/cen.12072
4. Kasturi K, Fernandes L, Quezado M, et al. Cushing disease in a patient with multiple endocrine neoplasia type 2B. Journal of Clinical and Translational Endocrinology Case Reports. 2017;4:1-4. doi: https://doi.org/10.1016/j.jecr.2017.02.001
5. Frederiksen A, Rossing M, Hermann P, et al. Clinical Features of Multiple Endocrine Neoplasia Type 4: Novel Pathogenic Variant and Review of Published Cases. Journal of Clinical Endocrinology and Metabolism. 2019,104(9):3637–3646. doi: https://doi.org/10.1210/jc.2019-00082
6. Scheithauer BW, Kovacs K, Horvath E, et al. Pituitary blastoma. Acta Neuropathologica. 2008;116:657–666. doi: https://doi.org/10.1007/s00401-008-0388-9
7. Chong AS, Han H, Albrecht S, et al. DICER1 syndrome in a young adult with pituitary blastoma. Acta Neuropathologica. 2021;142(6):1071-1076. doi: https://doi.org/10.1007/s00401-021-02378-0
8. Cazabat L, Bouligand J, Salenave S, et al. Germline AIP Mutations in Apparently Sporadic Pituitary Adenomas: Prevalence in a Prospective Single-Center Cohort of 443 Patients. Journal of Clinical Endocrinology and Metabolism. 2012;97(4):E663–E670. doi: https://doi.org/10.1210/jc.2011-2291
9. Hernández-Ramírez LC, Tatsi C, Lodish MB, et al. Corticotropinoma as a component of carney complex. Journal of the Endocrine Society. 2017;1(7):918-925. doi: https://doi.org/10.1210/js.2017-00231
10. Loughrey PB, Baker G, Herron B, et al. Invasive ACTH-producing pituitary gland neoplasm secondary to MSH2 mutation. Cancer Genetics. 2021;256-257:36-39. doi: https://doi.org/10.1016/j.cancergen.2021.03.008
11. Bengtsson D, Joost P, Aravidis C, et al. Corticotroph pituitary carcinoma in a patient with lynch syndrome (LS) and pituitary tumors in a Nationwide LS cohort. Journal of Clinical Endocrinology and Metabolism. 2017;102(11):3928-3932. doi: https://doi.org/10.1210/jc.2017-01401
12. Theodoropoulou M, Reincke M, Fassnacht M, Komada M. Decoding the genetic basis of Cushing’s disease: USP8 in the spotlight. European Journal of Endocrinology. 2015;173: M73–M83. doi: https://doi.org/10.1530/EJE-15-0320
13. Wanichi IQ, Mariani BMP, Frassetto FP. Cushing’s disease due to somatic USP8 mutations: a systematic review and metaanalysis. Pituitary. 2019;22(4):435-442. doi: https://doi.org/10.1007/s11102-019-00973-9
14. Faucz FR, Tirosh A, Tatsi C, et al. Somatic USP8 gene mutations are a common cause of pediatric Cushing disease. Journal of Clinical Endocrinology and Metabolism. 2017;102(8):2836-2843. doi: https://doi.org/10.1210/jc.2017-00161
15. Cohen M, Persky R, Stegemann R, et al. Germline USP8 mutation associated with pediatric Cushing disease and other clinical features: a new syndrome. Journal of Clinical Endocrinology and Metabolism. 2019;104(10):4676-4682. doi: https://doi.org/10.1210/jc.2019-00697
16. Theodoropoulou M, Reincke M. Genetics of Cushing’s disease: from the lab to clinical practice. Pituitary. 2022;25:689–692. doi: https://doi.org/10.1007/s11102-022-01253-9
17. Treppiedi D, Di Muro G, Marra G, et al. USP8 inhibitor RA-9 reduces ACTH release and cell growth in tumor corticotrophs. Endocrine-Related Cancers. 2021;28(8):573-582. doi: https://doi.org/10.1530/ERC-21-0093
18. Simon J, Theodoropoulou M. Genetics of Cushing’s disease. Journal of Neuroendocrinology. 2022;e13148. doi: https://doi.org/10.1111/jne.13148
19. Sbiera S, Perez-Rivas LG, Taranets L, et al. Driver mutations in USP8 wild-type Cushing’s disease. Neuro Oncology. 2019;21(10):1273–1283. doi: https://doi.org/10.1093/neuonc/noz109
20. Uzilov AV, Taik P, Cheesman KhC. USP8 and TP53 drivers are associated with CNV in a corticotroph adenoma cohort enriched for aggressive tumors. The Journal of Clinical Endocrinology & Metabolism. 2021;106(3):826–842. doi: https://doi.org/10.1210/clinem/dgaa853
21. Clynes D, Jelinska C, Xella B, et al. Suppression of the alternative lengthening of telomere pathway by the chromatin remodeling factor ATRX. Nature Communication. 2015;6:7538. doi: https://doi.org/10.1038/ncomms8538
22. Molenaar JJ, Koster J, Zwijnenburg DA, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012;483(7391):589-593. doi: https://doi.org/10.1038/nature10910
23. Fishbein L, Khare S, Wubbenhorst B, et al. Whole exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nature Communications. 2015;6:6140. doi: https://doi.org/10.1038/ncomms7140.
24. Job S, Draskovic I, Burnichon N, et al. Telomerase activation and ATRX mutations are independent risk factors for metastatic pheochromocytoma and paraganglioma. Clinical Cancer Research. 2019;25(2):760-770. doi: https://doi.org/10.1158/1078-0432.CCR-18-0139
25. Singhi AD, Liu TC, Roncaioli JL, et al. Alternative lengthening of telomeres and loss of DAXX/ATRX expression predicts metastatic disease and poor survival in patients with pancreatic neuroendocrine tumors. Clinical Cancer Research. 2017;23(2):600-609. doi: https://doi.org/10.1158/1078-0432.CCR-16-1113.
26. Casar-Borota O, Botling J, Granberg D, et al. Serotonin, ATRX, and DAXX expression in pituitary adenomas: markers in the differential diagnosis of neuroendocrine tumors of the sellar region. American Journal of Surgical Pathology. 2017;41(9):1238-1246. doi: https://doi.org/10.1097/PAS.0000000000000908
27. Casar-Borota O, Boldt HB, Engstrom BE, et al. Corticotroph aggressive pituitary tumors and carcinomas frequently harbor ATRX mutations. The Journal of Clinical Endocrinology & Metabolism. 2020;20(20):1–12. doi: https://doi.org/10.1210/clinem/dgaa749
28. Vetrivel Sh, Zhang R, Engel M, et al. Circulating microRNA Expression in Cushing’s Syndrome. Frontiers in Endocrinology. 2021;12. doi: https://doi.org/10.3389/fendo.2021.620012
29. Малыгина А.A., Белая Ж.Е., Никитин А.Г., и соавт. Экспрессии микрорнк в плазме крови, оттекающей от гипофиза, у пациентов с болезнью Иценко–Кушинга и АКТГ-эктопированным синдромом // Проблемы эндокринологии. — 2021. — Т. 67. — №6. — С. 18-30. doi: https://doi.org/10.14341/probl12817
30. Belaya Zh, Khandaeva P, Nonn L, et al. Circulating Plasma microRNA to Differentiate Cushing’s Disease From Ectopic ACTH Syndrome. Frontiers in Endocrinology. 2020;11:331. doi: https://doi.org/10.3389/fendo.2020.00331
31. Garbicz F, Mehlich D, Rak B. Increased expression of the microRNA 106b~25 cluster and its host gene MCM7 in corticotroph pituitary adenomas is associated with tumor invasion and Crooke’s cell morphology. Pituitary. 2017;20:450–463. doi: https://doi.org/10.1007/s11102-017-0805-y
32. Peculis R, Niedra H, Rovite V. Large scale molecular studies of pituitary neuroendocrine tumors: novel markers, mechanisms and translational perspectives. Cancers. 2021;13: 1395. doi: https://doi.org/10.3390/cancers13061395
33. Neou M, Villa C, Armignacco R, et al. Pangenomic classification of pituitary neuroendocrine tumors. Cancer Cell. 2020,37(1):123-134.e5. doi: https://doi.org/10.1016/j.ccell. 2019.11.002
34. Salomon MP, Wang X, Marzese DM, et al. The epigenomic landscape of pituitary adenomas reveals specific alterations and differentiates among acromegaly, Cushing’s disease and endocrine-inactive subtypes. Clinical Cancer Research. 2018;24(17):4126–4136. doi: https://doi.org/10.1158/1078-0432.CCR-17-2206
Лапшина Анастасия Михайловна, к.м.н.
117036, Москва, ул. Дм. Ульянова, д. 11
|
|
1. Рисунок 1. Мультиомиксные методы изучения кортикотропином. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(352KB)
|
Метаданные ▾ | |
Лапшина А.М. Молекулярно-генетические аномалии в кортикотропных опухолях гипофиза: фундаментальные исследования и перспективы использования в клинической практике. Проблемы Эндокринологии. 2024;70(3):23-30. https://doi.org/10.14341/probl13273
Lapshina A.M. Molecular genetic abnormalities in ACTH-secreting pituitary tumors (corticotropinomas): fundamental research and prospects for use in clinical practice. Problems of Endocrinology. 2024;70(3):23-30. (In Russ.) https://doi.org/10.14341/probl13273

 |
![]()
![]()

![]()
117292, Российская Федерация, Москва, ул. Дм. Ульянова, д.11



































