




Содержание
Перейти к:
https://doi.org/10.14341/probl13519
Перейти к:
Врожденный изолированный дефицит адренокортикотропного гормона (ВИДА) — орфанное аутосомно-рецессивное заболевание, обусловленное патогенными вариантами в гене ТВХ19 (1q24.2). В статье представлено описание двух клинических случаев с классической манифестацией ВИДА в неонатальном периоде, подтвержденного генетически, причем в одном из них выявлен впервые описанный вариант в гене TBX19. Диагноз установлен на 8-м и 22-м месяцах жизни, несмотря на появление клинически значимых симптомов в периоде новорожденности у обеих пациенток. Клинические проявления гипогликемии присутствовали у обеих пациенток: у пациентки №2 — с первых суток жизни (эпизод апноэ), у пациентки №1 — с 7 месяцев (судороги). У пациентки №1 основными проявлениями заболевания были холестатическая желтуха, гепатомегалия, признаки цитолиза гепатоцитов, нарушение белковосинтетической функции печени, что может свидетельствовать о развитии неинфекционного холестатического гепатита. Связь поражения печени с гипокортизолемией подтверждают улучшение и постепенная нормализация клинико-лабораторных изменений на фоне терапии гидрокортизоном. У пациентки №2 признаки холестаза отсутствовали. Лабораторно отмечались низкие уровни кортизола при сниженном или низконормальном — адренокортикотропного гормона (АКТГ), что подтверждает центральный гипокортицизм. При молекулярно-генетическом исследовании у обеих пациенток обнаружены патогенные варианты в гене ТВХ19 в гомозиготном состоянии: у пациентки №1 c.82C>T(p.Q28X), у пациентки №2 — c.469-1G>A, ранее не описанный.
Скородок Ю.Л., Кожевникова А.В., Плотникова Е.В., Иоффе И.Ю., Тюльпаков А.Н. Два случая врожденного изолированного дефицита адренокортикотропного гормона вследствие патогенных вариантов в гене TBX19. Проблемы Эндокринологии. 2025;71(3):51-55. https://doi.org/10.14341/probl13519
Skorodok Yu.L., Kozhevnikova A.V., Plotnikova E.V., Ioffe I.Y., Tiulpakov A.N. Two cases of congenital isolated adrenocorticotropic hormone deficiency due to pathogenic variants in TBX19. Problems of Endocrinology. 2025;71(3):51-55. (In Russ.) https://doi.org/10.14341/probl13519
Врожденный изолированный дефицит АКТГ (ВИДА) — орфанное аутосомно-рецессивное заболевание, обусловленное патогенными вариантами в гене ТВХ19 (MIM 604614), 1q24.2. Фактор транскрипции T-box (TPIT), кодируемый ТВХ19, необходим для терминальной дифференцировки клеток гипофиза, продуцирующих проопиомеланокортин, а также экспрессии гена POMC [1]. Заболевание с одинаковой частотой поражает мальчиков и девочек, в 40–42% отмечаются близкородственные браки [2][3]. В большинстве случаев ВИДА манифестирует в неонатальном периоде тяжелыми гипогликемиями, часто с приступами судорог и обструктивной желтухой [4]. Неспецифичность симптомов и низкая частота заболевания могут привести к поздней диагностике ВИДА [5][6], в связи с чем риск летального исхода в неонатальном периоде достигает 20–25% [3][7].
Представляем двух пациенток с ВИДА с классической манифестацией в неонатальном периоде (желтухой у первой, гипогликемией и апноэ у второй), подтвержденным генетически, причем у одной из них выявлен впервые описанный вариант в гене TBX19.
Ребенок от второй физиологической беременности (1 беременность — медицинский аборт), родилась доношенной с массой тела 3750 г, длиной 52 см, малыми аномалиями развития: кранио-фациальные дисморфии и левосторонняя косолапость. Брак близкородственный: родители — двоюродные брат и сестра.
Неонатальный скрининг на врожденный гипотиреоз, врожденную гиперплазию коры надпочечников, галактоземию, фенилкетонурию, муковисцидоз — отрицательный.
В возрасте 1,5 месяца обследована в связи с затяжной желтухой и плохой прибавкой массы тела; отмечалась гипербилирубинемия (общий билирубин — 91 мкмоль/л; норма 8,5–20,5), гиперферментемия (АЛТ — 182,2 Ед/л; норма 5–30, АСТ — 144,5 Ед/л; норма 8–40, ГГТП — 80,8 Ед/л; норма 10–60), гипогликемия без клинических проявлений (глюкоза крови 2,2–2,7 ммоль/л; норма 3,3–5,5). При УЗИ выявлена гепатомегалия, при эластографии — фиброз печени легкой степени. Подтверждено наличие цитомегаловирусной инфекции (повышенный уровень Anti-CMV-IgG, положительная ПЦР на CMV), проведена противовирусная терапия без существенного эффекта.
Пациентка в 7 месяцев экстренно госпитализирована в связи с судорогами. Наблюдались нарушение сознания (сопор), мышечная гипотония, брадикардия (ЧСС 110 уд./мин.) и брадипноэ (ЧД 10 в 1 мин), артериальная гипотония (АД 65/40 мм рт.ст.), иктеричность кожных покровов, гепатоспленомегалия (печень +5,0 см, селезенка +2,0 см из-под реберной дуги), ахоличный стул, темная моча. Отмечалась высокорослость (длина тела — 73 см; SDS 2,03) при дефиците веса (масса тела — 7300 г, SDS ИМТ -2,38). В ходе обследования выявлены тяжелая гипогликемия (глюкоза крови — 0,6 ммоль/л; норма 3,3–5,5), гиперферментемия (АЛТ — 400 Ед/л; норма 5–30, АСТ — 180 Ед/л; норма 8–40), гипербилирубинемия (общий билирубин — 200 мкмоль/л; норма 8,5–20,5, непрямой — 142 мкмоль/л; норма 3,4–12), гипопротеинемия (общий белок — 53 г/л; норма 56–79), анемия (Hb — 97 г/л; норма 114–140, эритроциты — 3,69×10¹²/л; норма 4–5,3), гипокоагуляция (коалиновое время — 97 сек.; норма 50–70, тромбиновое время — 20 сек; норма — 15–18, ПТИ — 63,2%; норма — 78–142, МНО — 1,45; норма — 0,85–1,25, АПТВ — 74 сек.; норма 35–45). Исключено инфицирование Toxoplasma, H. simplex 1, 2, Mycoplasma hominis/pneumoniae, Chlamidia trahomatis/pneumoniae, HBV, HCV, VIH (методом ИФА), ЕВV, CMV, HНV (ПЦР), наследственные болезни обмена: аминоацидопатии, органические ацидемии, дефекты β-окисления жирных кислот (тандемная масс-спектрометрия).
При УЗИ подтверждена гепатоспленомегалия на фоне диффузных изменений паренхимы печени, холестаза.
В процессе дифференциальной диагностики сочетания персистирующей гипогликемии и неинфекционной желтухи выявлены сниженный уровень кортизола (15,9 нмоль/л; норма 138–635) при низком АКТГ (4,99 пг/мл; норма 8,17–46,32). Гиперинсулинизм, нарушение других тропных функций аденогипофиза и гипотиреоз исключены (табл. 1). Это позволило диагностировать ВИДА. Учитывая тяжесть состояния, назначена интенсивная терапия гидрокортизоном парентерально в дозе 75 мг/м²/сут. с последующим постепенным снижением под контролем клинических симптомов и гликемии до поддерживающей 10 мг/м²/сут. в таблетированном виде.
Таблица 1. Уровни гормонов и глюкозы крови пациентки №1 в 7 месяцев
|
Показатель |
Результат |
Норма |
|
ТТГ, мМЕ/л |
3,52 |
0,62–8,0 |
|
свТ4, пмоль/л |
12,9 |
10–26 |
|
СТГ, нг/мл |
3,5 |
1,3–9,1 |
|
ИФР-1, нг/мл |
29,2 |
28–131 |
|
Инсулин, мкЕд/мл |
0,2 |
2,3–26 |
|
С-пептид, нг/мл |
0,1 |
0,36–3,6 |
|
Глюкоза, ммоль/л |
0,6 |
3,3–5,5 |
Примечание. ТТГ — тиреотропный гормон, свТ4 — свободный Т4, СТГ — соматотропный гормон, ИФР-1 — инсулиноподобный фактор роста — 1.
При МРТ визуализированы арахноидальная киста левых отделов цистерны продолговатого мозга, диффузная корково-подкорковая атрофия больших полушарий головного мозга.
Для установления причины заболевания проведено секвенирование по Сэнгеру гена TBX19: у пациентки обнаружен ранее описанный [8] патогенный вариант (NM_005149.2.): c.82C>T(p.Q28X) в гомозиготном состоянии, у матери — в гетерозиготном состоянии (рис. 1 б, в).
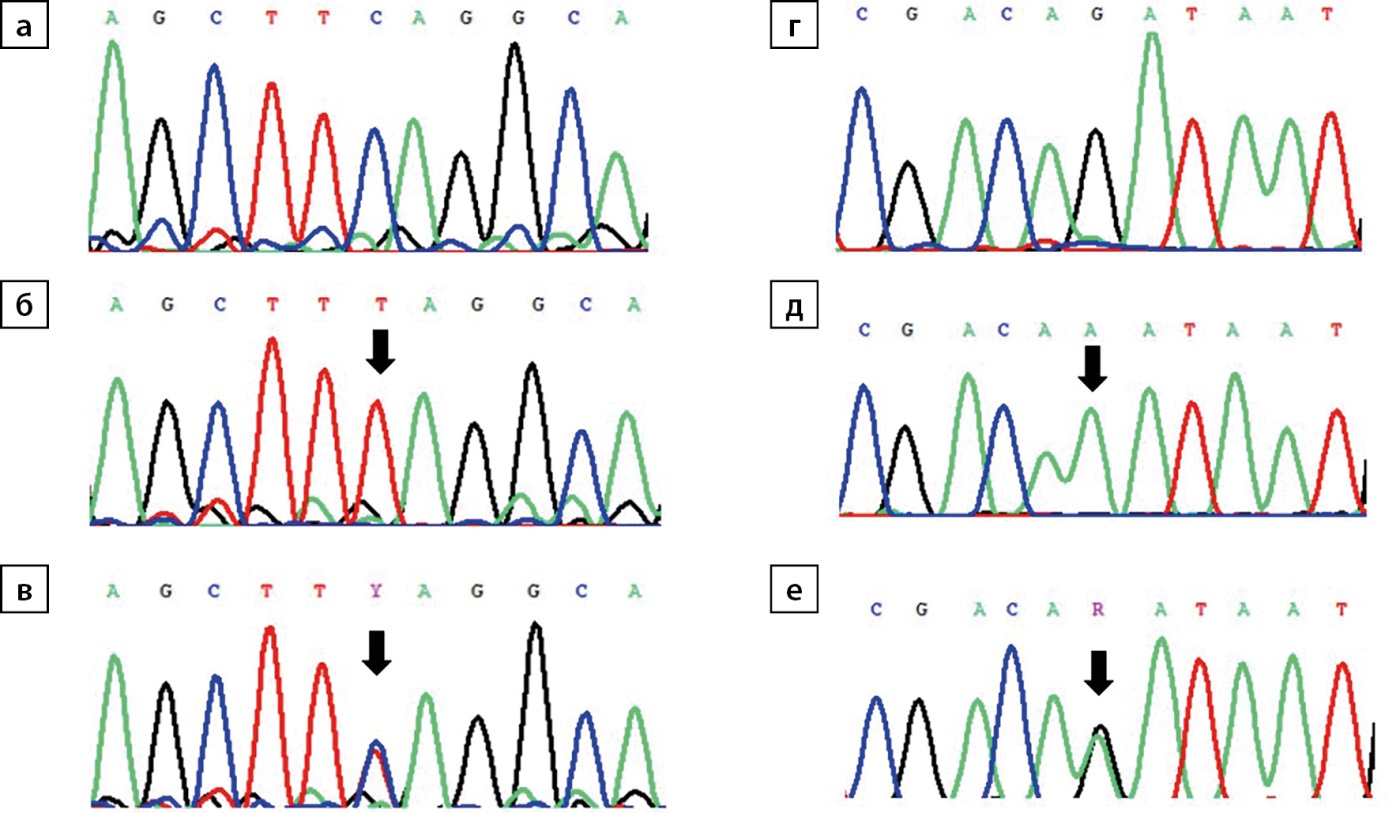
Рисунок 1. Хроматограммы с фрагментами сиквенса гена TBX19 (позиции с выявленным вариантом указаны стрелкой):
а) фрагмент экзона 1 NM_005149.2:c.79_87 (последовательность дикого типа); б) гомозиготная транзиция C>T в положении NM_005149.2:c.82 (пациентка 1); в) гетерозиготная транзиция C>T в положении NM_005149.2:c.82 (мать пациентки 1); г) фрагмент стыка интрон 2/экзон 3 NM_005149.2:c.469-6_473 (последовательность дикого типа); д) гомозиготная транзиция G>A в положении NM_005149.2:c.469-1 (пациентка 2); е) гетерозиготная транзиция G>A в положении NM_005149.2:c.469-1 (отец пациентки 2).
На 5-й день терапии устранена гипогликемия (глюкоза — 4,1 ммоль/л), через 2 месяца разрешилась желтуха, снизилась активность ферментов печени (АЛТ=125 Ед/л, АСТ=93,3 Ед/л, ГГТП=65,1 МЕд/л), через 3 месяца сохранялись ускоренные темпы роста на фоне нормализации массы тела (длина тела — 79 см, SDS +2,57, масса тела — 9200 г, SDS по росту +0,16).
Пациентка родилась от пятой беременности, протекавшей на фоне токсикоза и хронической фетоплацентарной недостаточности (старший брат 11 лет — здоров), вторых родов на 38-й неделе с массой тела 3340 г, длиной 52 см. Брак неродственный. В первые сутки отмечался эпизод апноэ, на вторые двукратно выявлена гипогликемия (глюкоза крови — 1,1 и 2,4 ммоль/л). В 2,5 месяца госпитализирована в неврологическое отделение в связи с повторным эпизодом апноэ и гипогликемическими судорогами (гликемия — 2,0 ммоль/л). Проведена КТ головного мозга, выявлены признаки смешанной гидроцефалии, субатрофии вещества головного мозга. Причина гипогликемии не установлена. Рекомендована вальпроевая кислота. В 3,5 месяца при плановом обследовании выявлены гипокортизолемия (кортизол — 27,6 нмоль/л; норма 138–635), умеренные гипертиреотропин- и гипотироксинемия (ТТГ — 8,9 мМЕ/л; норма 0,62–8,0, свТ4 — 9,05 пмоль/л; норма 11,5–20,4). В 4 месяца отмечалась высокорослость (длина тела — 68 см; SDS 3,1) при дефиците веса (масса тела — 6400 г, SDS ИМТ -2,6). В 7,5 месяцев госпитализирована в неврологическое отделение в связи с приступом тонико-клонических судорог на фоне поствакцинальной лихорадки. В ходе обследования неоднократно отмечалась гипогликемия (минимальная глюкоза крови — 0,59 ммоль/л). Диагностированы инфекционно-аллергический энцефалит (тяжелая форма), симптоматическая эпилепсия. В возрасте 1 года 9 месяцев повторно выявлена гипокортизолемия (кортизол — 24,4 нмоль/л) при низконормальном АКТГ 3,68 пг/мл (норма 0–30), диагностирован центральный гипокортицизм, начата терапия гидрокортизоном 15 мг/м²/сут. Подтвержден субклинический гипотиреоз (ТТГ — 16,3 мМЕ/л, свТ4 — 14,3 пмоль/л), назначен левотироксин — 12,5 мкг/сут.
В 2 года 1 месяц госпитализирована для контроля терапии и уточнения диагноза с жалобами на задержку психомоторного развития: начала ходить после года, не произносит слов. На фоне терапии гидрокортизоном в прежней дозе сохраняется высокорослость (рост — 98 см, SDS 3,8) и высокая скорость роста (19,1 см/год; SDS 3,9), масса тела нормализовалась (вес — 17 кг, SDS ИМТ 0,9); достигнута нормогликемия (глюкоза — 3,66–4,01 ммоль/л). Несмотря на нерегулярный прием левотироксина в минимальной дозе субклинический гипотиреоз не прогрессирует (ТТГ — 10,6 мМЕ/л; норма 0,64–5,76, свТ4 — 13,2 пмоль/л; норма 11,5–20,4). Проведено секвенирование по Сэнгеру гена TBX19, у пациентки обнаружен ранее не описанный в HGMD патогенный вариант (NM_005149.2.): c.469-1G>A в гомозиготном состоянии, у обоих родителей — в гетерозиготном состоянии (рис. 1 д, е).
ВИДА вследствие патогенных вариантов в гене ТВХ19 крайне редкое заболевание, данные по заболеваемости и распространенности ограничены. В мировой литературе в основном освещаются единичные клинические случаи, которых насчитывается не более 80. Впервые доказательства связи ВИДА с дефектами в гене ТВХ19 были представлены в 2001 г. Lamolet с соавт., показавшими коэкспрессию генов Tpit и Pomc в клеточных линиях гипофиза мыши, и выявившими мутации в гене TPIT (TBX19) у 2 пациентов с вторичной надпочечниковой недостаточностью [9]. Abali Z.Y. и соавт. (2019 г.) обобщили данные о 66 ранее описанных пациентах, среди которых у большинства симптомы развивались в неонатальном периоде и заболевание диагностировалось до 2 лет [2][3][8].
Первые симптомы заболевания неспецифичны, что значительно затрудняет диагностику ВИДА [8]. Наиболее частыми симптомами считают гипогликемию (100%), в том числе симптоматическую (судороги, 53%), холестатическаую желтуху (62%), а также гипотермию или лихорадку, шок [3]. По данным ряда авторов, один из первых симптомов — приступ апноэ на 1–11-й дни жизни [1][10][11]. У некоторых пациентов развивается особая форма гепатита — неонатальный неинфекционный гепатит, связанный с гипокортизолемией; диагноз подтверждается при биопсии почти в каждом втором случае сочетания холестатической желтухи и гепатомегалии [8][11].
И в наших случаях диагноз установлен только на 8-м и 22-м месяцах жизни, несмотря на появление клинически значимых симптомов в периоде новорожденности у обеих пациенток, хотя гипогликемические судороги у пациентки №1 развились лишь к 7 месяцам, а у пациентки №2 отсутствовали признаки холестаза. Эпизод апноэ в первые сутки жизни у пациентки №2 следует расценить как результат гипогликемии. У пациентки №1 основными проявлениями заболевания были холестатическая желтуха (ахоличный стул, темная моча, гипербилирубинемия), признаки цитолиза гепатоцитов (гиперферментемия), нарушение белковосинтетической функции печени (гипоальбуминемия, гипокоагуляция), гепатомегалия. И хотя биопсию печени не проводили, данные изменения и отсутствие эффекта от противовирусной терапии могут свидетельствовать о развитии неинфекционного холестатического гепатита. Связь поражения печени с гипокортизолемией подтверждают улучшение и постепенная нормализация клинико-лабораторных изменений на фоне терапии гидрокортизоном. Учитывая вышесказанное, целесообразно при наличии гипогликемии ± холестаза у новорожденного или младенца исключить гипокортицизм.
Данные о росте пациентов с ВИДА вследствие патогенных вариантов в гене ТВХ19 противоречивы. Некоторые авторы описывают высокорослость на фоне нормального или сниженного ИФР-1 с нормализацией темпов роста на фоне заместительной терапии глюкокортикоидами [2][4][6]. Другие исследователи не выявили изменений антропометрических показателей у пациентов [12][13]. У обеих описываемых пациенток отмечалась высокорослость на фоне дефицита веса: умеренной степени к 7 месяцам (пациентка №1) и тяжелой в возрасте 4 месяцев (пациентка №2). Нормальный уровень СТГ и низконормальный — ИФР1 у пациентки №1 исключают связь высокорослости с нарушением в системе ГР-ИФР1. В отличие от литературных данных, высокие темпы роста на фоне терапии в течение 3–4 месяцев сохранялись при нормализации массы тела. Ввиду невозможности исследования уровня ИФР1-связывающего белка 5 подтвердить или опровергнуть гипотезу о связи высокорослости с отсутствием ингибирования этого белка при дефиците глюкокортикоидов, обусловленном ВИДА, не удалось [2].
Ряд авторов сообщали о различных дисморфологических признаках у пациентов с ВИДА, таких, как порок Арнольда-Киари 1-го типа и черепно-лицевые малые аномалии [3][4][6]. У пациентки №1 отмечались тригоноцефалия, скошенный затылок, узкие глазные щели, эпикант, антимонголоидный разрез глаз, широкая плоская переносица, микростомия, готическое небо, низко посаженные уши, левосторонняя косолапость. У пациентки №2 подобные аномалии не наблюдались. Установить связь фенотипа с патогенным вариантом гена ТВХ19 не представляется возможным. У обеих пациенток при визуализации выявлена диффузная атрофия/субатрофия вещества головного мозга, у пациентки №1 дополнительно — арахноидальная киста, у пациентки №2 — гидроцефалия. Подобных изменений ЦНС в литературе не описано, хотя в некоторых случаях отмечали умственную отсталость. Исключить связь субатрофии/атрофии вещества головного мозга у наших пациенток с тяжелыми гипогликемиями не представляется возможным.
Для ВИДА характерны резко сниженный уровень кортизола при сниженном [2][3][4][7][8][10][12] или нормальном [1][6] АКТГ. Следует отметить, что оценка уровня АКТГ зависит от чувствительности тест-систем. У наших пациенток отмечались аналогичные результаты: низкие уровни кортизола при сниженном или низконормальном АКТГ.
Среди пациентов с ВИДА описано несколько случаев идиопатического субклинического гипотиреоза [5][6][7][9]. В последующем уровни ТТГ нормализовались, в том числе спонтанно или на фоне лечения левотироксином в минимальных дозах. У пациентки №2 также была выявлена гипертиреотропинемия (в 3,5 месяца), сохраняющаяся в 2 года 1 месяц на фоне нерегулярного приема левотироксина 12,5 мкг/сут.
По данным на 2021 г., в HGMD (http://www.hgmd.cf.ac.uk) включены 29 патогенных вариантов в гене TBX19: ряд миссенс-мутаций, большие и малые делеции, четыре мутации в пределах сайта сплайсинга и новая синонимичная мутация NM_005149.3:c.288G>A (p.T96=) [1].
У пациентки №1 обнаружен патогенный вариант c.82C>T(p.Q28X) в гомозиготном состоянии, ранее выявленный Vallette-Kasic S и соавт., 2005 [8], что позволило подтвердить диагноз «ВИДА». Авторы описали сходные проявления у родных брата и сестры с данной мутацией: гипогликемию с судорогами, холестатическую желтуху, однако гепатит подтвержден не был и не отмечены какие-либо малые аномалии развития.
У пациентки №2 выявлен ранее не описанный вариант в гене ТВХ19 c.469-1G>A в гомозиготном состоянии. Хотя в настоящее время данный вариант отсутствует в референтных популяционных базах данных (https://gnomad.broadinstitute.org/), он может быть расценен как патогенный и каузальный, так как относится к влияющим на сплайсинг и присутствует в гомозиготном состоянии у пациента с классической клинико-лабораторной картиной ВИДА.
Представленные нами клинические случаи подчеркивают, что при наличии персистирующей гипогликемии, особенно в сочетании с холестазом, у новорожденного или младенца необходимо исключить гипокортицизм.
Выявление врожденного изолированного дефицита АКТГ (двукратно низкий уровень кортизола в сочетании с неповышенным АКТГ при отсутствии нарушения других функций аденогипофиза) требует молекулярно-генетического исследования.
В одном из представленных в настоящей публикации случаев выявлен ранее не описанный вариант в гене ТВХ19 (NM_005149.2.): c.469-1G>A.
Источники финансирования. Молекулярно-генетическое исследование выполнено при частичном содействии Фонда поддержки и развития филантропии «КАФ».
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи.
Участие авторов. Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
Согласие пациента. Пациенты добровольно подписали информированное согласие на публикацию персональной медицинской информации в обезличенной форме в журнале «Проблемы эндокринологии».
Благодарности. Выражаем благодарность Иванову Дмитрию Владимировичу, врачу педиатру, за своевременный диагностический поиск и оказание квалифицированной экстренной врачебной помощи пациентке №1.
1. Maudhoo A, Maharaj A, Buonocore F, et al. Missplicing due to a synonymous, T96= exonic substitution in the T-box transcription factor TBX19 resulting in isolated ACTH deficiency. Endocrinol Diabetes Metab Case Reports. 2021;2021. doi: https://doi.org/10.1530/EDM-21-0128
2. Abali ZY, Yesil G, Kirkgoz T, et al. Evaluation of growth and puberty in a child with a novel TBX19 gene mutation and review of the literature. Hormones (Athens). 2019;18(2):229-236. doi: https://doi.org/10.1007/s42000-019-00096-7
3. Couture C, Saveanu A, Barlier A, et al. Phenotypic homogeneity and genotypic variability in a large series of congenital isolated ACTH-deficiency patients with TPIT gene mutations. J Clin Endocrinol Metab. 2012;97(3):E486-E495. doi: https://doi.org/10.1210/jc.2011-1659
4. Kardelen Al AD, Poyrazoğlu Ş, Aslanger A, et al. A Rare Cause of Adrenal Insufficiency - Isolated ACTH Deficiency Due to TBX19 Mutation: Long-Term Follow-Up of Two Cases and Review of the Literature. Horm Res Paediatr. 2019;92(6):395-403. doi: https://doi.org/10.1159/000506740
5. Vieira IH, Mourinho Bala N, Ramos F, et al. A serious and unusual presentation of congenital isolated ACTH deficiency due to TBX19 mutation, beyond the neonatal period. Endocrinol Diabetes Metab Case Rep. 2022. doi: https://doi.org/10.1530/EDM-22-0277
6. Weijing K, Liping Z, Tiantian Z, et al. A Case of Congenital Isolated Adrenocorticotropic Hormone Deficiency Caused by Two Novel Mutations in the TBX19 Gene. Front Endocrinol (Lausanne). 2019;10:251. 2019. doi: https://doi.org/10.3389/fendo.2019.00251
7. Peng C, Sun G, Tang Z, Hou X. Congenital Isolated ACTH Deficiency Caused by TBX19 Gene Mutation: A Family Report. Front Pediatr. 2020;7:546. 2020. doi: https://doi.org/10.3389/fped.2019.00546
8. Vallette-Kasic S, Brue T, Pulichino AM, et al. Congenital isolated adrenocorticotropin deficiency: an underestimated cause of neonatal death, explained by TPIT gene mutations. J Clin Endocrinol Metab. 2005;90(3):1323-1331. doi: https://doi.org/10.1210/jc.2004-1300
9. Lamolet B, Pulichino AM, Lamonerie T, et al. A pituitary cell-restricted T box factor, Tpit, activates POMC transcription in cooperation with Pitx homeoproteins. Cell. 2001;104(6):849-859. doi: https://doi.org/10.1016/s0092-8674(01)00282-3
10. Li SJ, Hu LY, Zhang R, et al. 新生儿急性心功能不全1例 [Acute heart failure in a neonate]. Zhongguo Dang Dai Er Ke Za Zhi. 2024;26(3):321-324. doi: https://doi.org/10.7499/j.issn.1008-8830.2311087
11. Alsaleem M, Saadeh L, Misra A, Madani S. Neonatal isolated ACTH deficiency (IAD): a potentially life-threatening but treatable cause of neonatal cholestasis. BMJ Case Rep. 2016;2016:bcr2016215032. 2016. doi: https://doi.org/10.1136/bcr-2016-215032
12. Akcan N, Serakıncı N, Turkgenc B, et al. A Novel TBX19 Gene Mutation in a Case of Congenital Isolated Adrenocorticotropic Hormone Deficiency Presenting with Recurrent Respiratory Tract Infections. Front Endocrinol (Lausanne). 2017;8:64. doi: https://doi.org/10.3389/fendo.2017.00064
13. Unal E, Yıldırım R, Taş FF, et al. A rare cause of neonatal hypoglycemia in two siblings: TBX19 gene mutation. Hormones (Athens). 2018;17(2):269-273. doi: https://doi.org/10.1007/s42000-018-0028-2
Скородок Юлия Леонидовна, к.м.н. кафедра детских болезней им проф. И. М. Воронцова ФП и ДПО, доцент, врач детский эндокринолог
Санкт-Петербург
Кожевникова Анжелика Владимировна, кафедра детских болезней им проф. И. М. Воронцова ФП и ДПО, ассистент, врач детский эндокринолог
194100, Санкт-Петербург, ул. Литовская, д. 2
Плотникова Елена Валерьевна, к.м.н. кафедра детских болезней им проф. И. М. Воронцова ФП и ДПО, доцент, врач детский эндокринолог
Санкт-Петербург
Тюльпаков Анатолий Николаевич, д.м.н., заведующий кафедрой генетики эндокринных болезней, врач детский эндокринолог
Москва
|
|
1. Рисунок 1. Хроматограммы с фрагментами сиквенса гена TBX19 (позиции с выявленным вариантом указаны стрелкой): а) фрагмент экзона 1 NM_005149.2:c.79_87 (последовательность дикого типа); б) гомозиготная транзиция C>T в положении NM_005149.2:c.82 (пациентка 1); в) гетерозиготная транзиция C>T в положении NM_005149.2:c.82 (мать пациентки 1); г) фрагмент стыка интрон 2/экзон 3 NM_005149.2:c.469-6_473 (последовательность дикого типа); д) гомозиготная транзиция G>A в положении NM_005149.2:c.469-1 (пациентка 2); е) гетерозиготная транзиция G>A в положении NM_005149.2:c.469-1 (отец пациентки 2). | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(705KB)
|
Метаданные ▾ | |
Скородок Ю.Л., Кожевникова А.В., Плотникова Е.В., Иоффе И.Ю., Тюльпаков А.Н. Два случая врожденного изолированного дефицита адренокортикотропного гормона вследствие патогенных вариантов в гене TBX19. Проблемы Эндокринологии. 2025;71(3):51-55. https://doi.org/10.14341/probl13519
Skorodok Yu.L., Kozhevnikova A.V., Plotnikova E.V., Ioffe I.Y., Tiulpakov A.N. Two cases of congenital isolated adrenocorticotropic hormone deficiency due to pathogenic variants in TBX19. Problems of Endocrinology. 2025;71(3):51-55. (In Russ.) https://doi.org/10.14341/probl13519

 |
![]()
![]()

![]()
117292, Российская Федерация, Москва, ул. Дм. Ульянова, д.11



































