


Clinical endocrinology
BACKGROUND: Hyperprolactinemia accompanies growth hormone hypersecretion in approximately 25-39% of cases. There is a recommendation to determine the level of prolactin in clinical guidelines for diagnosis and treatment of acromegaly. However, there is no understanding of the necessity to investigate the IGF-1 level in patients with hyperprolactinemia and a pituitary adenoma.
AIM: Determining the proportion of patients with hyperprolactinemia and pituitary adenoma, who were examined for IGF-1 levels, and identifying the proportion of patients with acromegaly among this cohort.
MATERIALS AND METHODS: Between December 2019 and December 2022 a single-center observational single-stage single-sample uncontrolled study was conducted. At the first stage of the study, the proportion of patients with pituitary adenoma and hyperprolactinemia with studied IGF-1 levels was determined, according to medical records. At the second stage of the study, patients without known indicators of IGF-1 were determined. The concentration of growth hormone was studied during the oral glucose load in the case of increased IGF-1 levels.
RESULTS: At the first stage, 105 patients were included in the study. The level of IGF-1 was determined in 41/105 (39%) cases. There were 22/41 (53.7%) cases in the subgroup with pituitary incidentalomas and 19/64 (29.7%) cases in the subgroup with hyperprolactinemia among them. At the second stage, the IGF-1 level was additionally determined in 53 patients with hyperprolactinemia and pituitary adenoma (total 94 patients). The level of IGF-1 was elevated in 11/94 patients, further acromegaly was confirmed in 3/94 patients (3.2%).
CONCLUSION: In real clinical practice the level of IGF-1 is studied only in 39% of cases in patients with pituitary adenoma and hyperprolactinemia. The disease was detected in 3 cases (3.2%) out of 94 people with hyperprolactinemia and pituitary adenoma without clinical manifestations of acromegaly. We consider the study of IGF-1 levels justified as a screening for acromegaly in patients with hyperprolactinemia and pituitary adenoma.
BACKGROUND: Knowledge of the physiological mechanisms of adaptation arising in response to changes in photoperiods is especially important for residents of the European North. In the literature, there is practically no information about photoperiodic dynamics of serum dopamine level, despite its significant role in the regulation of the body’s activity. The mutual modulating effect of the dopaminergic and thyroid systems is known.
AIM: To show the ratio of dopamine levels and the content of hormones, protines and autoantibodies of the thyroid system, taking into account photoperiod of the year, in practically healthy populations of the European North.
MATERIALS AND METHODS: Healthy male population (20 men) of Arkhangelsk was examined in various photoperiods of the year (80 samples): an increase in the length of daylight hours (March), its maximum duration (June), a decrease (September), and a minimum duration (December). The inhabitants of the settlements and the nomadic aboriginal population (100 men) were examined during 2 photoperiods of the year — March and December. The serum levels of iodothyronines, TSH, TG, antibodies to TPO, antibodies to TG and plasma level of dopamine were determined using ELISA methods.
RESULTS: Residents of Arkhangelsk in June compared to December have higher levels of dopamine (0.502 and 0.365 nmol/l, p=0.01), T3 (1.09 and 0.94 nmol/l, p=0.003), T4 (113.45 and 99.03 nmol/l, p=0.0002). In September, compared with June, a decrease in dopamine (0.235 nmol/l, p=0.0003), T3 (0.92 nmol/l, p=0.004) was recorded with an increase in T4/T3 ratio from 106.54 to 117.89 units (p=0.006). The nomadic aboriginal population in March compared with December showed a tendency to a higher content of dopamine (0.00 and 0.394 nmol/l, p=0.07) with the decrease in fT4 (15.20 and 13.90, p=0.015), fT4/fT3 ratio from 3.13 to 2.28 units (p=0.006). In December, 67% of nomadic population had undetectable dopamine values (0 nmol/l) and 22% — excess dopamine values, in March 27% — excess values.
CONCLUSION: Unidirectional changes in dopamine and thyroid activity in men of the European North were shown with their decrease during periods of decrease and minimum daylight hours and an increase during periods of increase and maximum daylight hours.
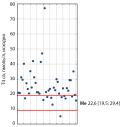
BACKGROUND: Thyrotropin-secreting pituitary adenomas (TSH-PA) are a rare cause of thyrotoxicosis and account for 0.5-2% of all pituitary adenomas. Taking into account the rarity of the disease, it is extremely important to analyze each case of TSH-PA. AIM: To analyze the clinical characteristics and treatment outcomes of patients with TSH-PA, as well as to determine preoperative and early postoperative factors that predict long-term remission.
MATERIALS AND METHODS: In a single-center retrospective study we analyzed clinical signs, laboratory and instrumental studies, as well as the treatment outcomes of patients with TSH-PA from 2010 to 2023. Preoperative factors, as well as TSH level measured on day 3 postoperatively, were evaluated for their ability to predict long-term remission when comparing groups of patients with and without remission. RESULTS: The study included 45 patients with TSH-PA (14 men, 31 women), with a median age of 45 years [30; 57]. The most common clinical manifestations of TSH-PA were: cardiac arrhythmia in 37 (82.2%) patients, thyroid pathology in 27 (60%), neurological disorders in 24 (53.35%). Most PAs were macroadenomas (n=35, 77.8%). Preoperatively, 28 (77.8%) patients received somatostatin analogs, and 20 (71.4%) patients were euthyroid at the time of surgery. Surgical treatment was performed in 36 (80%) patients, postoperative remission was achieved in 31 cases (86.1%). Administration of somatostatin analogues to patients with no remission/relapse after surgery lead to the remission in 100% of cases (4/4). A 1 mm increase in PA size raised the odds of recurrence/no remission by 1.15-fold,and PA invasion during surgery — by 5.129 fold. A TSH level on day 3 postoperatively above 0.391 mIU/L (AUC, 0.952; 95% CI 0.873–1.000; standard error 0.04; p<0.001) identifies patients with relapse/absence of remission after surgical treatment (sensitivity = 100%, specificity = 88.9%).
CONCLUSION: The TSH-PA in the structure of PAs is extremely rare, and as a result, most of them are misdiagnosed and detected already at the stage of macroadenoma. The most effective method of treatment is transnasal transsphenoidal adenomectomy. Somatostatin analogues can be used as second-line therapy if surgical treatment is ineffective. We have proposed a possible model for postoperative TSH levels (>0.391 mU/l) to predict recurrence of TSH-PA, which requires validation on an expanded number of cases.
Currently, all pheochromocytoma/paraganglioma (PPGLs) are considered malignant due to metastatic potential. Consequently, PPGLs are divided into «metastatic» and «non-metastatic». Metastatic PPGLs can be with synchronous metastasis (metastases appear simultaneously with the identified primary tumor) or metachronous (metastases develop after removal of the primary tumor). The term metastatic PPGLs is not used in the presence of tumor invasion into surrounding organs and tissues, without the presence of distant metastases of lymphogenic or hematogenic origin. It is generally believed that about 10% of pheochromocytomas and about 40% of sympathetic paragangliomas have metastatic potential. On average, the prevalence of PPGLs with the presence of metastases is 15–20%. Risk factors for metastatic PPGLs are widely discussed in the literature, the most significant of which are groups of clinical, morphological and genetic characteristics. The review presents a discussion of such risk factors for metastatic PPGLs as age, localization and type of hormonal secretion of the tumor, the size and growth pattern of the adrenal lesion, the presence of necrosis and invasion into the vessels, the tumor capsule surrounding adipose tissue, high cellular and mitotic activity, Ki-67 index, expression of chromogranin B and S100 protein, the presence of genetic mutations of three main clusters (pseudohypoxia, kinase signaling and Wnt signaling).
Over the past two decades, a number of authors have proposed various predictor factors and scales for assessing a probability of metastatic PPGLs. The review contains detailed description and comparison of sensitivity and specificity of such predictor scales as PASS, GAPP, M-GAPP, ASES and COPPS.

BACKGROUND: Pheochromocytoma (PHEO) is a tumor from the chromaffin tissue of the adrenal medulla, capable of hyperproduction of catecholamines. The increased production of hormones by the tumor leads to catecholamine crises, which have a pathological effect on all organs and systems. In the primary diagnosis of pheochromocytomas, it is important to determine the level of the metabolite of catecholamines — metanephrines. Currently, in clinical practice, various methods are used to determine the level of this metabolite: in blood plasma or in urine, total or only free form, fractionated analysis or unfractionated.
AIM: Comparison of the effectiveness of various methods for determining the level of metanephrines for the diagnosis of pheochromocytomas.
MATERIALS AND METHODS: A retrospective single-center cohort study was conducted on a sample of patients who were initially operated on for adrenal neoplasm at the Pirogov St. Petersburg State University High Medical Technology Clinic from November 2007 to December 2022 and who passed analysis to determine the level of blood or urine metanephrins before surgical treatment. The results of tests for metanephrine and tumor size were evaluated.
RESULTS: 1088 patients with adrenal neoplasms who underwent surgical treatment were examined, of which 348 had histologically confirmed the presence of pheochromocytoma. Four types of metanephrine assays were compared: free fractionated plasma metanephrines (232 patients), unfractionated daily urine metanephrines (431 patients), fractionated total daily urine metanephrines (427 patients) and fractionated free daily urine metanephrines (178 patients). The greatest sensitivity was demonstrated by the analysis of free fractionated plasma methanephrines (95.4%). Unlike others, the sensitivity of this analysis did not decrease in the group of patients with small pheochromocytomas (3 cm or less). The greatest specificity was demonstrated by the analysis of unfractionated metanephrines in daily urine (97.8%), with the lowest sensitivity among all tests (67.6%). The study of fractionated total daily urine metanephrins showed good results of sensitivity and specificity, only slightly inferior to the best indicators, and the analysis of free daily urine metanephrins demonstrated unexpectedly low efficiency. There is a positive correlation between the level of metanephrine in the blood and the size of the tumor.
CONCLUSION: Based on the data obtained, the preferred assays for the primary diagnosis of pheochromocytoma can be considered the determination of fractionated free plasma metanephrines and fractionated total daily urine metanephrines, which is consistent with relevant clinical recommendations. It was found that the size of the tumor correlates with the severity of an increase in the level of metanephrins determined by any of the described methods.

We presented the clinical case of neurofibromatosis type 1 (NF-1) associated with pheochromocytoma (PHEO) in a man under 40 years old without family history. The diagnosis of NF-1 was established based on 4 signs of the disease (multiple café au lait macules, scoliotic changes in posture, the presence of multiple neurofibromas, Lisch nodules). The diagnosis of PHEO was determined by a significant increase of free metanephrin/normethanephrin levels in daily urine, a malignant CT phenotype of the right adrenal tumor, and confirmed by pathomorphological study. Genetic tests revealed a new mutation in one of the alleles of NF1 gene, a deletion of a 566 bp gene fragment, including exon 19 with a size of 73 bp. This mutation leads to splicing of exons 18 and 20, frameshift, and termination of protein synthesis. A study of the level of transcription of the genes associated with PHEO (RET, TMEM127, MAX, FGFR, MET, MERTK, BRAF, NGFR, Pi3, AKT, MTOR, KRAS, MAPK) was conducted, a statistically significant decrease in the level of transcription of the KRAS and BRAF genes and increase in the level of transcription of the TMEM127 gene in comparison with control samples have been detected. This case demonstrates the need for timely recognition of NF-1 for further appropriate patient’s follow up and show the effectiveness of a multidisciplinary approach to the diagnosis and treatment of NF-1-associated catecholamine-secreting tumors.

Polydipsia is a pathologically increased thirst, satisfied by the intake of water in large quantities, which can manifest itself in various somatic or mental diseases and at first glance is similar to a true vasopressin deficiency. Central diabetes insipidus (CDI) is a disease of the hypothalamic-pituitary region characterized by the inability of the kidneys to reabsorb water and concentrate urine, which is based on a defect in the synthesis or secretion of vasopressin and is manifested by severe thirst and excretion of large amounts of hypotonic urine. The prevalence of the disease in the population is 1:25,000, which characterizes it as a fairly rare pathology of the hypothalamic-pituitary region. The peak incidence is between 30 and 40 years of age. According to various literary sources, the disease is not characterized by gender differences in prevalence, however, on the example of the Moscow population, women prevailed in the incidence structure of CND in a ratio of 2.2:1. Insidiousness, with apparent absences in the difficulty of diagnosing primary polydipsia, lies in the manifestations of water intoxication, thus this condition requires knowledge of clear diagnostic criteria by healthcare professionals and an interdisciplinary approach in the treatment of this condition. On the example of this clinical case, we will try to highlight the differential diagnostic criteria for psychogenic polydipsia in comparison with the true deficiency of arginine vasopressin (AVP) or central diabetes insipidus (CDI), which can be applied in real clinical practice.
Carbohidrates metabolism disturbancies
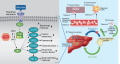
Breast cancer (BC) is a serious disease and is considered an important health problem worldwide. The prevalence of the disease in women according to Rosstat was 64,951 cases in the Russian Federation in 2020 (21.7% among all types of cancer). Hormone-dependent estrogen receptor-positive (HR+), human epidermal growth factor receptor type 2 negative (HER2-) metastatic breast cancer (mBC) accounts for 70% of all cases. About 40% of patients with ER+/HER2- mBC have mutations in the PIK3CA gene, leading to hyperactivation of the alpha isoform (p110α) of phosphatidylinositol 3-kinase (PI3K). Hormonal therapy with or without cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitor is considered the standard treatment for patients with ER+/HER2- mBC. However, acquired resistance to this therapy remains a problem. Innovative methods for the treatment of breast cancer are the use of targeted therapeutic agents aimed at direct inhibition of the PI3K pathway in combination with hormone therapy. Alpelisib is a PI3Kα-specific inhibitor. Hyperglycemia is the most common side effect of alpelisib treatment. Currently, there is a consensus on the prevention and correction of hyperglycemia in patients receiving therapy with alpelisib, which recommends that before starting therapy, in order to diagnose carbohydrate metabolism disorders and assess the risk of developing hyperglycemia, determine in all patients: the level of glycated hemoglobin (HbA1c), glucose fasting plasma (FPG), body mass index (BMI). And also to evaluate such risk factors as the presence of a family history of type 2 diabetes mellitus (DM 2), the presence of gestational diabetes in the patient’s history, or the fact of the birth of children weighing more than 4 kilograms.
Recently, new combinations of drugs have been actively used to treat disorders of carbohydrate metabolism, such as pioglitazone + metformin. This paper discusses the mechanism of action of PI3K inhibitors, new therapeutic combinations and their undesirable effects, and presents therapeutic experience.
Pediatric Endocrinology
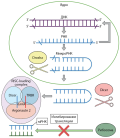
DICER1 syndrome is a rare genetic disorder with the progressive development of malignant and non-malignant diseases in childhood. The cause of this syndrome is a dusfunction of the endoribonuclease DICER, which plays an important role in the processing of microRNAs with subsequent regulation of the control of the expression of oncogenes and tumor suppressor genes. Clinical manifestations of dyseropathies is very different and may include both endocrine manifestations – multinodular goiter, differentiated thyroid cancers, ovarian stromal tumors, pituitary blastoma, and non–endocrine formations — pleuropulmonary blastoma, cystic nephroma, pineoblastoma. The presence of somatic mutations of the DICER1 gene is a resultant stage in the pathogenesis of dyseropathies, determining the further path of oncogenesis. At present, DICER1 syndrome is diagnosed extremely rarely, which leads to late detection of the components of the disease in the patient, late diagnosis of neoplasms, lack of family counseling. Diagnosis at the early stages of the disease, the development of screening programs for the management of these patients allows minimizing the risks of developing more malignant, aggressive forms of the disease.

Wiedemann-Rautenstrauch syndrome (neonatal progeroid syndrome) is an ultra-orphan disease from the group of premature aging syndromes with an autosomal recessive type of inheritance associated with mutations in the POLR3A, POLR3B, and POLR3GL genes encoding RNA polymerase III. The incidence of the disease is currently unknown. We present the first clinical description in Russian Federation of a patient 7 years 6 months old with Wiedemann-Rautenstrauch syndrome (compound heterozygous mutations in POLR3A gene) with progeroid features, adentia, growth retardation (height SDS -3,41, height velocity SDS -2,47), underweight (BMI SDS -6,20), and generalized lipodystrophy. The article presents the observation of the patient for 1.5 years, the world experience of dynamic follow-up of patients with neonatal progeroid syndrome, differential diagnosis, as well as recommendations for the management of patients with this syndrome. Given the lack of specific treatment to date, patients are observed by a multidisciplinary team of physicians.
IgG4-related disease is a rare chronic pathology manifested by lymphoplasmacytic infiltration of one or more organs, the formation of storiform fibrosis, tissue edema, and an increase of IgG4 in the blood. This disease was singled out as an independent nosological unit only in 2001. The incidence is less than 1 in 100,000 people per year. Almost any organ can be affected in IgG4-related disease. The association of Riedel's thyroiditis with IgG4 was established in 2010. Riedel's thyroiditis is an extremely rare inflammatory disease of the thyroid gland, which diagnosis is complicated by an atypical course and the absence of characteristic symptoms. Less than 300 clinical cases of the disease have been described in the world, only two from them were in children. This article presents a clinical case of a 6-year-old boy with Riedel's thyroiditis.
Clinical guidelines

On March 28, 2024, the Council of Experts “High-dose vitamin D (Devilam) in the practice of obstetrician-gynecologist, gynecologist and endocrinologist” was held in Moscow with the participation of leading experts gynecologists, endocrinologists and obstetricians-gynecologists, during which new possibilities for the use of high-dose vitamin D in patients of various ages who need correction of existing vitamin D deficiency or insufficiency.

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND 4.0).
ISSN 2308-1430 (Online)






































