


Editorial notice
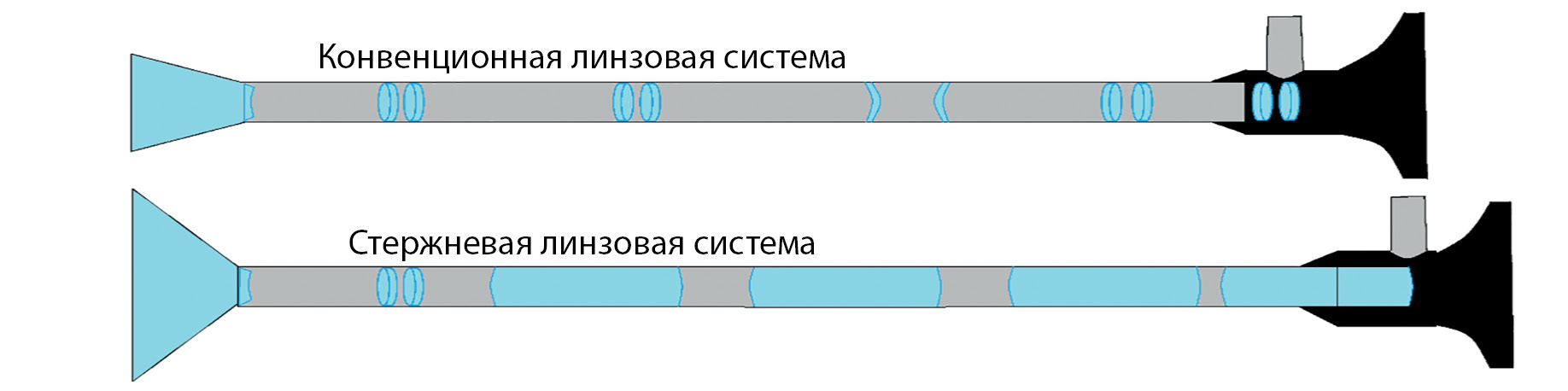
Currently, the treatment of pituitary adenomas is inextricably linked with transsphenoidal neurosurgical intervention. Modern technologies used in surgery for this pituitary pathology, such as endoscopy using angled optics, as well as the use of specialized instruments, sealing and hemostatic materials, increase the effectiveness of surgical treatment of pituitary adenomas and reduce the incidence of intraand postoperative complications. The development of radiation methods of diagnostics, such as MRI, makes it possible to more accurately identify the formation of the pituitary gland, assess its size, direction of growth, and the degree of invasion of surrounding tissues. The authors of the article described in detail the modern technique of transsphenoidal removal of pituitary adenoma using an endoscope. Each stage of the operation is described step by step, taking into account various anatomical features and illustrated. This article discusses the MRI characteristics of pituitary adenomas: size of the tumor, the direction of its growth, the degree of invasion of the cavernous sinuses, the compression effect on the structures of the chiasmal-sellar region. The use of treatment methods, knowledge of the features of MRI diagnostics described in this article greatly increase the effectiveness of the treatment of patients with pituitary adenomas and reduce the risk of complications after neurosurgical intervention in such patients.
Clinical endocrinology
Searching for aging key points is one of the main problems in geriatrics. More and more research in recent years has been devoted to the study of geroprotective mechanisms, the impact of various conditions and diseases on aging in general. Of particular importance is the determination of age-related involutive processes in the human body, whether they are part of normal aging or a condition that needs to be corrected to improve the functioning of organs and systems. An important mechanism of aging starts is a change in hormonal activity of endocrine glands, in particular in hormonal activity of thyroid. Frequency of hypothyroidism in advanced age explains relevance of the chosen topic. The aim of the review was to find out the role hypothyroidism in aging. The main task was to define, whether thyroid hormones decrease in older age was a protective factor or pathological process. A review of the literature over the past 10 years on subclinical treatment was carried out and we identified the most pressing issues associated with hypothyroidism and aging. We studied data on the relationship between hypothyroidism and major geriatric syndromes, with special attention paid to cognitive diseases and emotional disorders.
BACKGROUND: Hodgkin’s lymphoma (HL) is one of the most common malignant lymphoproliferative diseases. Chemotherapy and radiotherapy used in the treatment of LH induce a number of toxic effects leading to dysfunction of endocrine system. Hormonal disorders in HL and their relationships with the therapy used remain to be clarified.
AIM: To assess disorders of the endocrine function of thyroid, parathyroid glands and gonads in HL survivors.
MATERIALS AND METHODS: Screening of endocrine dysfunction of the thyroid, parathyroid glands and gonads was performed in 160 adult patients with HL, 55 men and 105 women, at remission stage induced by chemotherapy or chemoradiotherapy. Forty healthy subjects, matched by age, were acted as control. The levels of TSH, T3, free T4, PTH, FSH, LH, free testosterone, dehydroepiandrosterone sulfate (DHEA-S), and sex-hormone binding globulin (SHBG) were measured in blood serum by ELISA. Bone mineral density (BMD) was assessed by DEXA.
RESULTS: Hypothyroidism (25%), hyperparathyroidism (15.6%) and hypogonadism (29% of men and 25.3% of women) were the most prevalent endocrine disorders in LH survivors. Hypothyroidism was significantly more common in patients after chemoradiotherapy than in those who received only chemotherapy (χ2=9.4, р=0.002). In patients with hyperparathyroidism, there were negative correlations between PTH levels and BMD in the lumbar spine (r=-0.74, p=0.00002) and in the femoral neck (r=-0.66, p=0.0003). Men with HL demonstrated lower free testosterone concentrations when compared to control (p=0.04); LH and FSH levels were elevated (p=0.0004 and p=0.04, respectively). In men with HL the levels of DHEA-S were reduced (p=0.0009). The increased SHBG concentrations were revealed in 13 (23.6%) men. Women of reproductive age with HL had higher levels of LH in the luteal phase (p=0.05) and FSH in the follicular phase (p=0.02) than controls.
CONCLUSION: The data indicate a high prevalence of the dysfunctions of thyroid, parathyroid glands, and gonads in HL survivors. Screening for endocrine disorders in these patients is highly recommended.
Functioning pituitary adenomas and pheochromocytomas/paragangliomas are rare in the general population. Pituitary adenomas occur in the familial setting in approximately 5% of cases, whereas pheochromocytomas/paragangliomas can be hereditary in 30–40% of cases. Hereditary syndromes associated with pituitary adenomas include multiple endocrine neoplasia types 1 and 4, familial isolated pituitary adenomas, and Carney complex. Hereditary syndromes associated with pheochromocytomas/paragangliomas and genes, mutations in which predispose to their development, are more numerous. The first clinical descriptions of the co-occurrence of pituitary adenoma and pheochromocytoma/paraganglioma in one patient date back to the mid 20th century, however delineating such a co-occurrence into a particular syndrome («3PAs» (pituitary adenoma, pheochromocytoma, paraganglioma)) was suggested only in 2015. To date, approximately 100 cases of such a co-occurrence have been described in the literature. Mutations in genes encoding subunits of succinate dehydrogenase complex II (SDHx) are revealed in the majority of cases, much less common are mutations in MAX, MEN1 and some other genes. This review summarizes the current information on the «3PAs» syndrome.
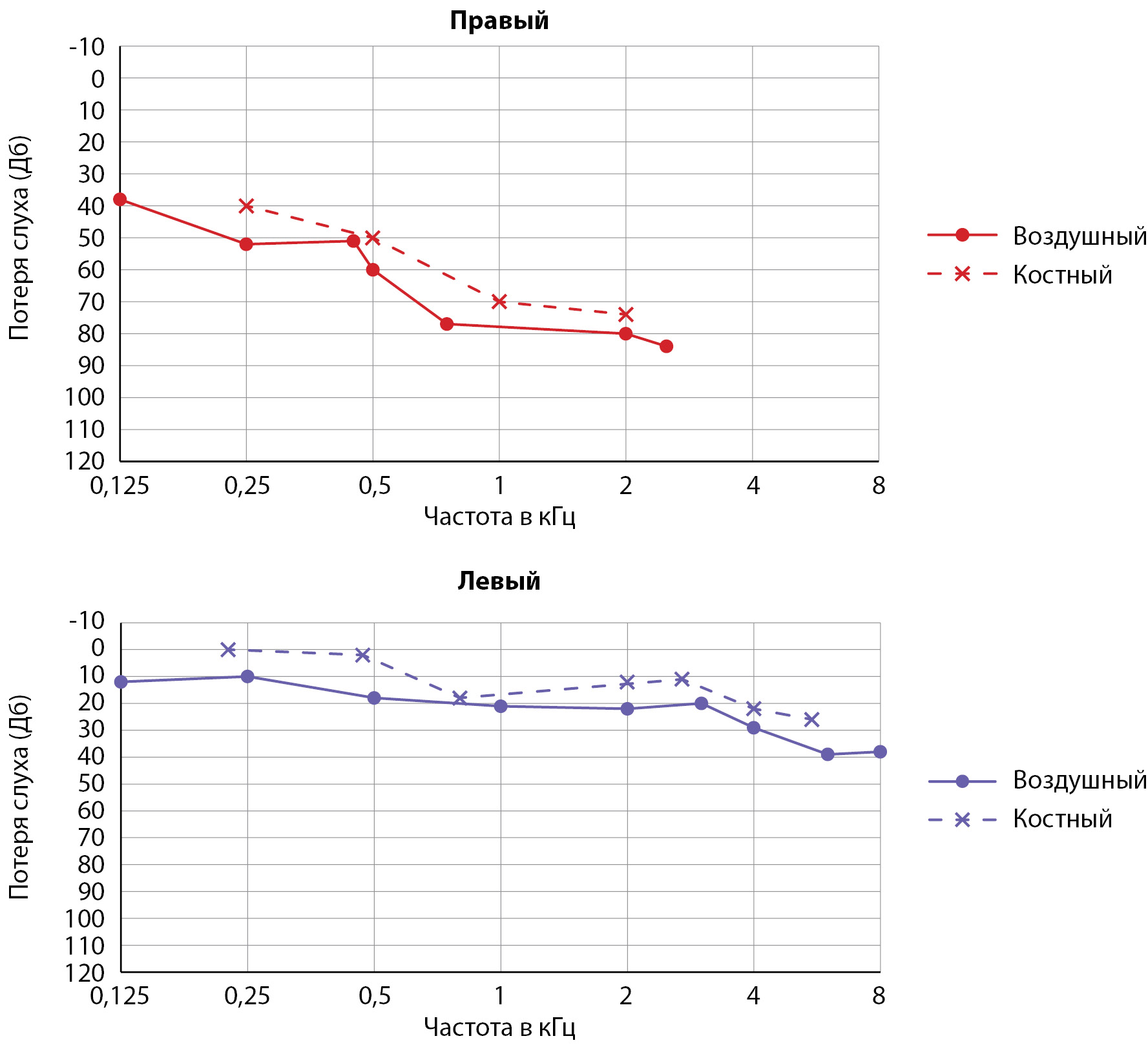
Acromegaly is a multifactorial neuroendocrine disease caused by hyperproduction of growth hormone (GH). In more than 95% of cases the reason of acromegaly the GH-secreting pituitary adenoma. In patients with this neuroendocrine disease, a slowly developing complex of symptom can manifest with concomitant pathological conditions, including auditory function disordersDiagnostic difficulties of acromegaly at the ambulatory stage determine the importance of doctor`s awareness in different medical specialties.
Here we demonstrate a clinical case of the improvement of the auditory function due to combined surgical and medical treatment of a patient with the pituitary macroadenoma, acromegaly and hearing loss.
Anamnesis features: a patient with an active stage of acromegaly and a pituitary macroadenoma measuring 57x35x32 mm with ante-, supra-, infra-, parasellar spread, (Knosp III(D), Knosp IV(S) noted a violation of auditory function. She was consulted by an otolaryngologist, sensorineural hearing loss on the right of the 3rd degree was diagnosed, on the left of the 1st degree. The patient underwent surgical treatment of pituitary adenoma, noted a significant improvement in auditory function in the early postoperative period. Six months later, repeated audiometry was performed, marked regression of hearing damage was noted.
The case described by us indicates the reversibility of a rare complication of acromegaly — hearing loss and the importance of an interdisciplinary approach in the management of patients with this pathology.
Carbohidrates metabolism disturbancies
BACKGROUND. The projected 68% increase in patients with type 2 diabetes mellitus (T2D) in the upcoming decades and the specific pathophysiological course of the disease are critical factors for the development of optimal disease management tactics in the Asian population. It is now known that β-cell dysfunction is dominant in the pathogenesis of T2D in Asians. In a number of Asian countries, incretin therapy is the leading therapy.
AIM. To review literature on glucagon-like peptide-1 (GLP-1) secretion and clinical trial results of GLP-1 receptor agonist class (GLP-1RA) drugs as well as to evaluate their effectiveness in Asian population with T2D.
MATERIALS AND METHODS. A review of studies on pathophysiological aspects of GLP-1 secretion and evaluation of the efficacy of therapy with GLP-1RA preparations registered and used in clinical practice in Asian regions.
RESULTS. Several studies in Asian countries have shown that intact GLP-1 levels were significantly lower in both T2D patients and healthy Japanese volunteers; as well as in patients with impaired glucose tolerance. It is suggested that either impaired secretion of GLP-1 in the gut, accelerated processing by dipeptidyl peptidase-4, or a combination of both are responsible for the decrease in GLP-1. The greater efficacy of GLP-1RA treatment in achieving glycemic control in Asian T2D patients was presented by Kim Y.G. et al. in a meta-analysis of 15 randomised controlled trials, the reduction in HbA1c on GLP-1RA treatment averaged -1.16% in Asian-dominated studies and -0.83% in non-Asian-dominated studies. In the PIONEER 9 clinical programme, similar results were obtained, with oral semaglutide having a more pronounced effect on glycaemic control in Japanese patients. Thus, the mean change in HbA 1c was -1.1%, 7 mg -1.5%, and 14 mg -1.7% at the 3 mg dose; whereas in the PIONEER 1 study in the global population, the mean change in HbA1c was -0.6%, -0.9% and -1.1% for 3, 7, 14 mg semaglutide, respectively. The PIONEER 10 study concluded that oral semaglutide was well tolerated by Japanese patients with T2D. Oral semaglutide reduced HbA1c (14 mg dose) and body weight (7 and 14 mg doses) more significantly compared to dulaglutide at 0.75 mg dose. Results of a pooled analysis of long-acting GLP-1RA showed a more significant reduction in cardiovascular event risk in the Asian subpopulation.
CONCLUSION. The presented review describes benefits in glycemic control as well as in the reduction of relative cardiovascular event risks with GLP-1RA treatment in the Asian population, which requires further in-depth research and implies optimal management tactics in patients with T2DM.
Bones & Adipose tissues diseases
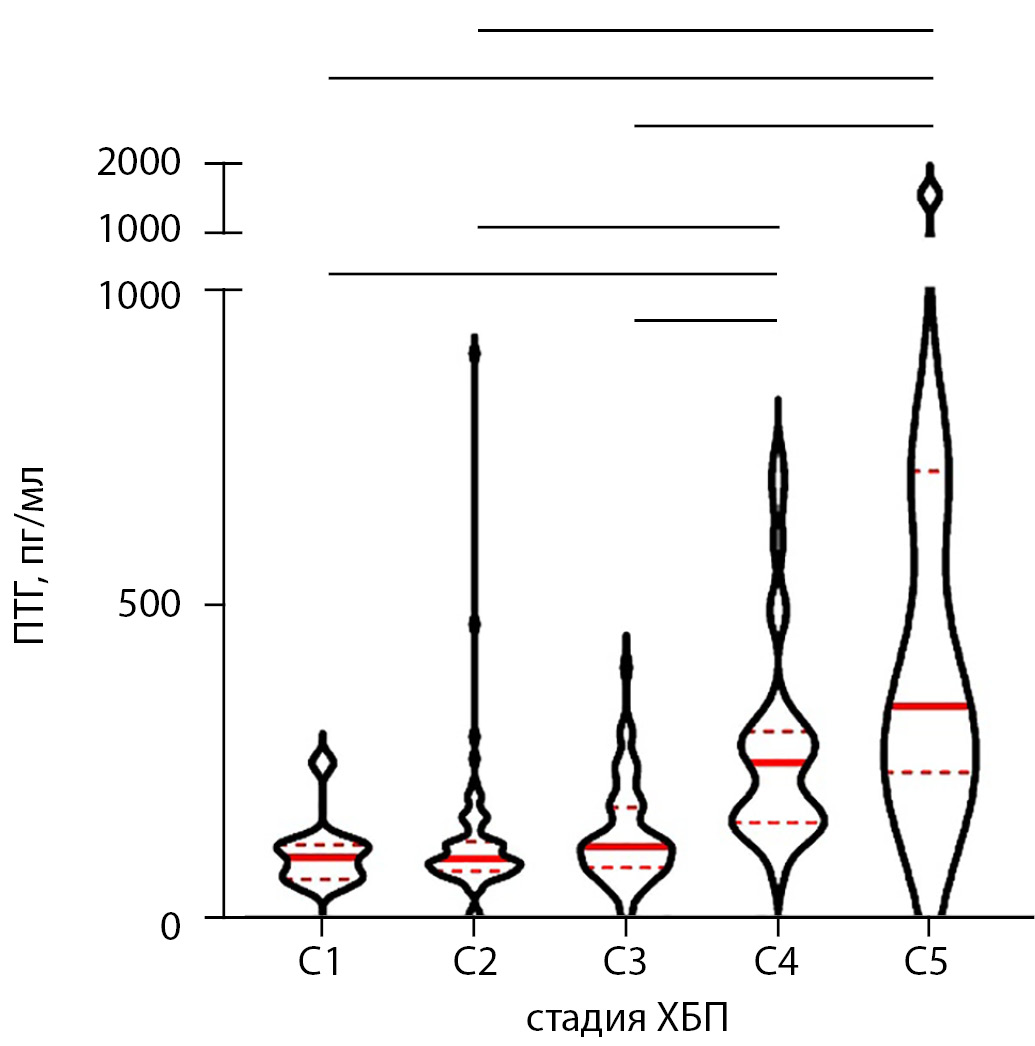
BACKGROUND: There is a lack of studies providing comprehensive data on the prevalence of mineral and bone disorders (MBD) laboratory abnormalities after kidney transplantation in Russia.
AIM: to obtain real-world data on the prevalence of the main mineral abnormalities among kidney transplant recipients and to revise their concomitant MBD therapy.
METHOD: This cross-sectional study included 236 patients with successful kidney transplantation. Their serum intact parathyroid hormone (iPTH), total calcium (Ca), phosphorus (P), and alkaline phosphatase (ALP) levels were measured.
RESULTS: Only 6.2% of our cohort had all laboratory parameters within the target range, whereas persistent HPT along with hypercalcemia was noted in almost one third of the patients (31%). Normal iPTH levels were observed in 13% cases; 84% of the patients had hyperparathyroidism. The fraction of patients with target iPTH did not differ between the groups with normal and decreased estimated glomerular filtration rate (eGFR) (p=0.118). Hypercalcemia was observed in 29% cases. The serum P level varied significantly in groups with different eGFR (p<0.0001), increasing with declining graft function. Furthermore, 40.7% of patients had ALP above the target range. While 123 patients received active vitamin D (alfacalcidol), 33 received monotherapy with inactive vitamin D (cholecalciferol). The control group consisted of 57 medication-naïve patients. The serum total Ca level varied significantly between the groups (p=0.0006), being higher in patients supplemented with cholecalciferol. The fraction of patients with normocalcemia was lowest in the cholecalciferol group (chi-square, р=0.0018).
CONCLUSION: The prevalence of biochemical abnormalities after kidney transplantation is high. Alfacalcidol usage may be safer than using cholecalciferol to prevent hypercalcemia development.
Pediatric Endocrinology
BACKGROUND: In 90% cases of girls and 25–60% cases of boys the cause of gonadotropin-dependent precocious puberty (PP) is unclear. Up to 25–27.5% of gonadotropin-dependent PP cases are monogenic and suggest autosomal-dominant inheritance with incomplete sex-dependent penetrance. To date, mutations in genes KISS1, KISS1R, MKRN3, DLK1 have been described as causal variants leading to precocious hypothalamic-pituitary axis activation in childhood. Genetic testing in patients with hereditary forms of PP can expand our knowledge of underlying molecular mechanisms of the disease and it is also necessary for genetic counselling.
AIM: To study clinical features and genetic characteristics of patients with idiopathic gonadotropin-dependent precocious puberty.
MATERIALS AND METHODS: A group of patients with idiopathic gonadotropin-dependent precocious puberty and positive family history (early or precocious puberty) was examined. Laboratory and instrumental diagnostic tests, full-exome sequencing (NGS, next-generation sequencing) were provided for all patients.
RESULTS: The study included 30 patients (29 girls, 1 boy) with idiopathic gonadotropin-dependent precocious puberty. The median of patients age at the time of the examination was 7,2 years [6,5; 7,7]. Positive family history presented in all cases: in 40% of patients on father’s side, in 37% — on mother’s side, in 23% of patients PP was diagnosed in siblings. The fullexome sequencing was conducted to 21 patients: in 61,9% of cases (95% CI [40;79]) nucleotide variants were identified in genes, associated with gonadotropin-dependent precocious puberty. MKRN3 gene defect was detected in most cases (77% cases (95% CI [49; 92]), which consistent with international data on its highest prevalence in the monogenic forms of PP. In 23% of cases (95% CI [7; 50]) nucleotide variants were identified in other candidate genes associated with neuroontogenesis and neuroendocrine regulation mechanisms of hypothalamic-pituitary axis.
CONCLUSION: Our study confirms that detailed family history data in children with PP provides a rational approach to molecular-genetic testing. Data of inheritance pattern and clinical manifestations will simplify the diagnosis of hereditary forms of disease and enhance genetic counselling of families, followed by timely examination and administration of pathogenetic therapy.
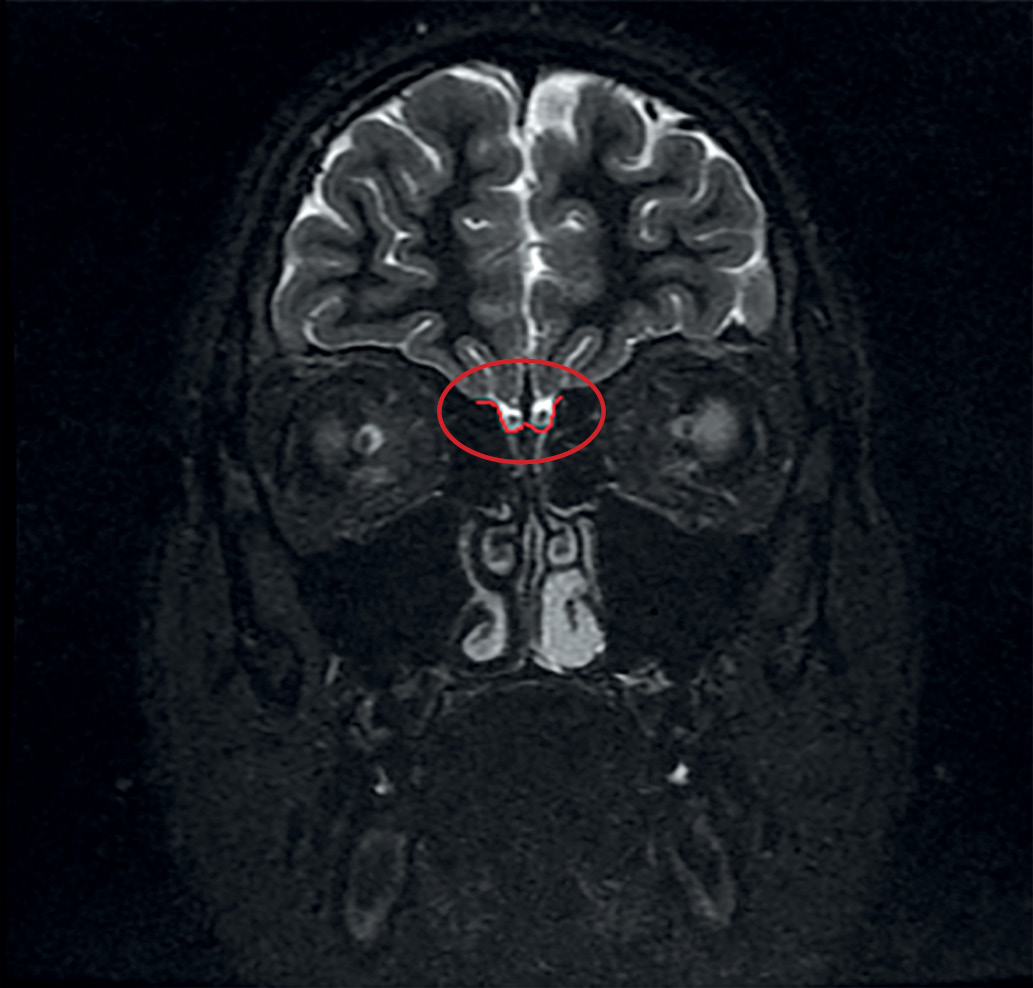
BACKGROUND: The majority of Kallmann patients have anosmia or hyposmia. This is how the disease is diagnosed. Some of them don’t have such complaints but olfactory dysfunction is diagnosed via olfactometry. Nowadays there is the lack of information about correlation between olfactometry results and subjective complaints. Correlation between olfactory bulbs size and olfactory dysfunction has been little studied.
AIM: To explore olfactory bulb size and olfactory function in patients with congenital isolated hypogonadotropic hypogonadism. To correlate olfactory bulb sizes and smell test scores.
MATERIALS AND METHODS: Single-centre comparative study. 34 patients were included. The main group consisted of 19 patients with hypogonadotropic (15 –with Kallmann syndrome, 4 — with normosmic hypogonadism). Olfactory bulbs MRI were provided to all the patients, olfactory test (Sniffin’ Sticks Test) and molecular-genetic studies were provided in all patients with hypogonadism. Control group consisted of 15 patients who were provided with orbits MRI. Olfactory bulbs were evaluated additionally in them.
RESULTS: Normal size of olfactory bulbs were only in 1 patient with hypogonadism. Olfactory bulbs height and width were significantly smaller in patients with hypogonadism in comparison with control group (p<0.01). Height median of right bulb was 1.0 mm [0.2; 1.8] in patients from the main group vs. 3.0 [2.5; 3.2] in controls, width median of right bulb was 1.0 mm [0.2; 1.9] in patients from the main group vs. 2.5 [2.0; 3.0] in controls. Height median of left bulb was 0.8 mm [0.0; 1.2] in patients from the main group vs. 3.0 [2.7; 3.2] in controls, width median of left bulb was 0.8 mm [0.0; 1.2] in patients from the main group vs. 2.5 [2.0; 3.0] in controls. Correlation has been established between left bulb height (r=0.59) and width (r=0.67) and olfactometry results (p<0.05). 4 patients had no anosmia complaints but had olfactory dysfunction according to Sniffin’ Sticks Tests.
CONCLUSION: Olfactometry was able to diagnose olfactory dysfunction in 78.5% (i.e. in 15 out of 19 patients with congenital isolated hypogonadotropic hypogonadism. However, anosmia complaints had only 11 out of 19 patients. It is the first results of olfactory bulb sizes in patients with hypogonadotropic hypogonadism in Russia. Uni — or bilateral hypoor aplasia were diagnosed in 94.7% patients with hypogonadism regardless of olfactory dysfunction. Bilateral olfactory bulbs hypoplasia were the most common MRI-finding (36.8%). Unilateral hypoor aplasia was diagnosed in 31.6% patients.
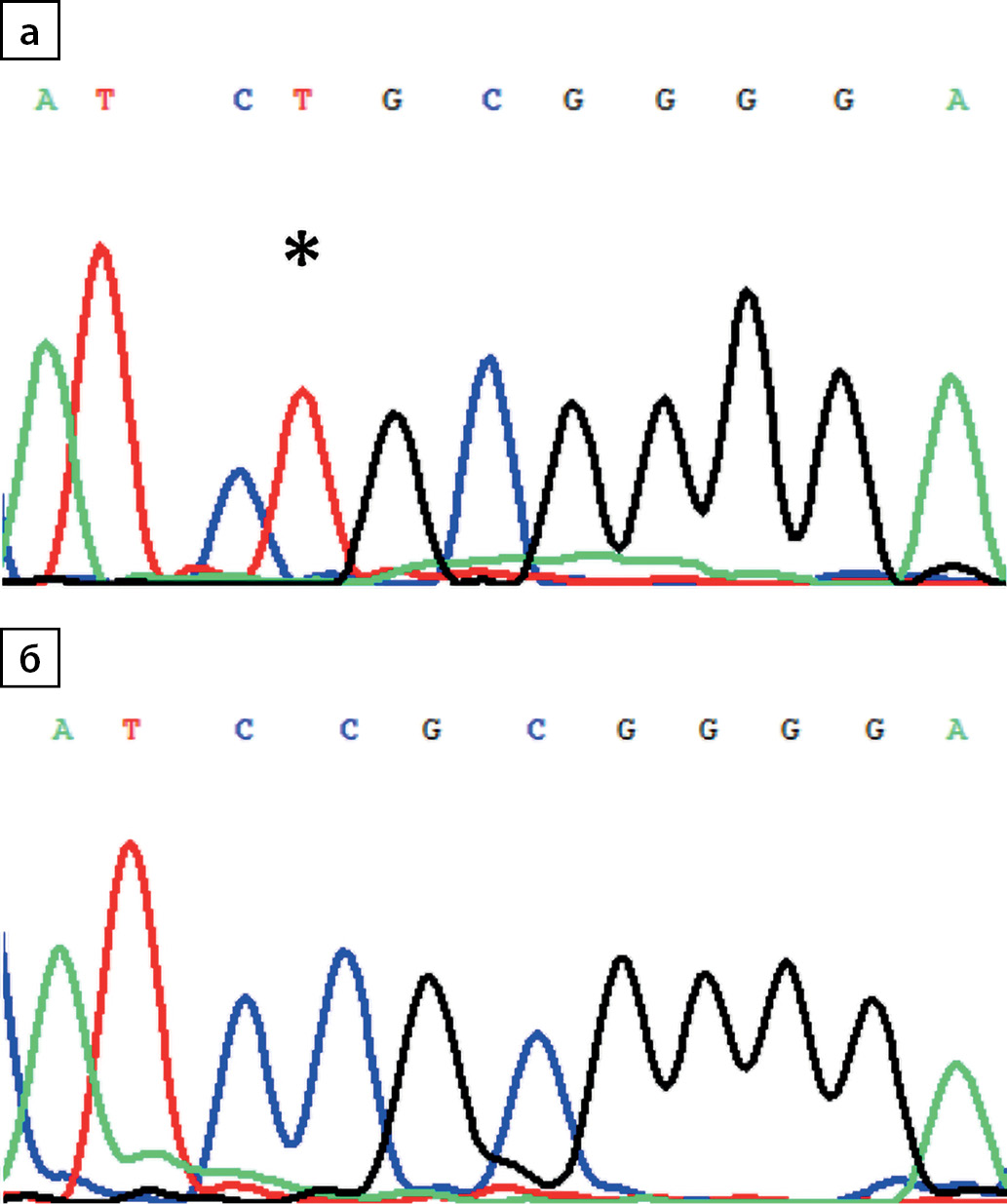
Congenital nephrogenic diabetes insipidus (CNDI, arginine vasopressin resistance) is a rare inherited disorder characterized by insensitivity of the kidney to the antidiuretic effect of vasopressin. NDI is clinically characterized by polyuria with hyposthenuria and nocturia and polydipsia. In the majority of cases, about 90%, nephrogenic diabetes insipidus is an X-linked recessive disorder caused by mutations in the AVP V2 receptor gene (AVPR2). In the remaining cases, about 10%, the disease is autosomal recessive or dominant and, for these patients, mutations in the aquaporin 2 gene (AQP2) have been reported. To date, the nucleotide variants registered in AQP2 were sporadic, there is no data on the presence of «frequent» mutations and the prevalence of the disease both among the global population and among individual ethnic groups. In this paper, we describe 12 cases of arginine vasopressin resistance caused by a new homozygous mutation p.R113C in AQP2 presented among the indigenous population of the Republic of Buryatia.
Reproductive Endocrinology
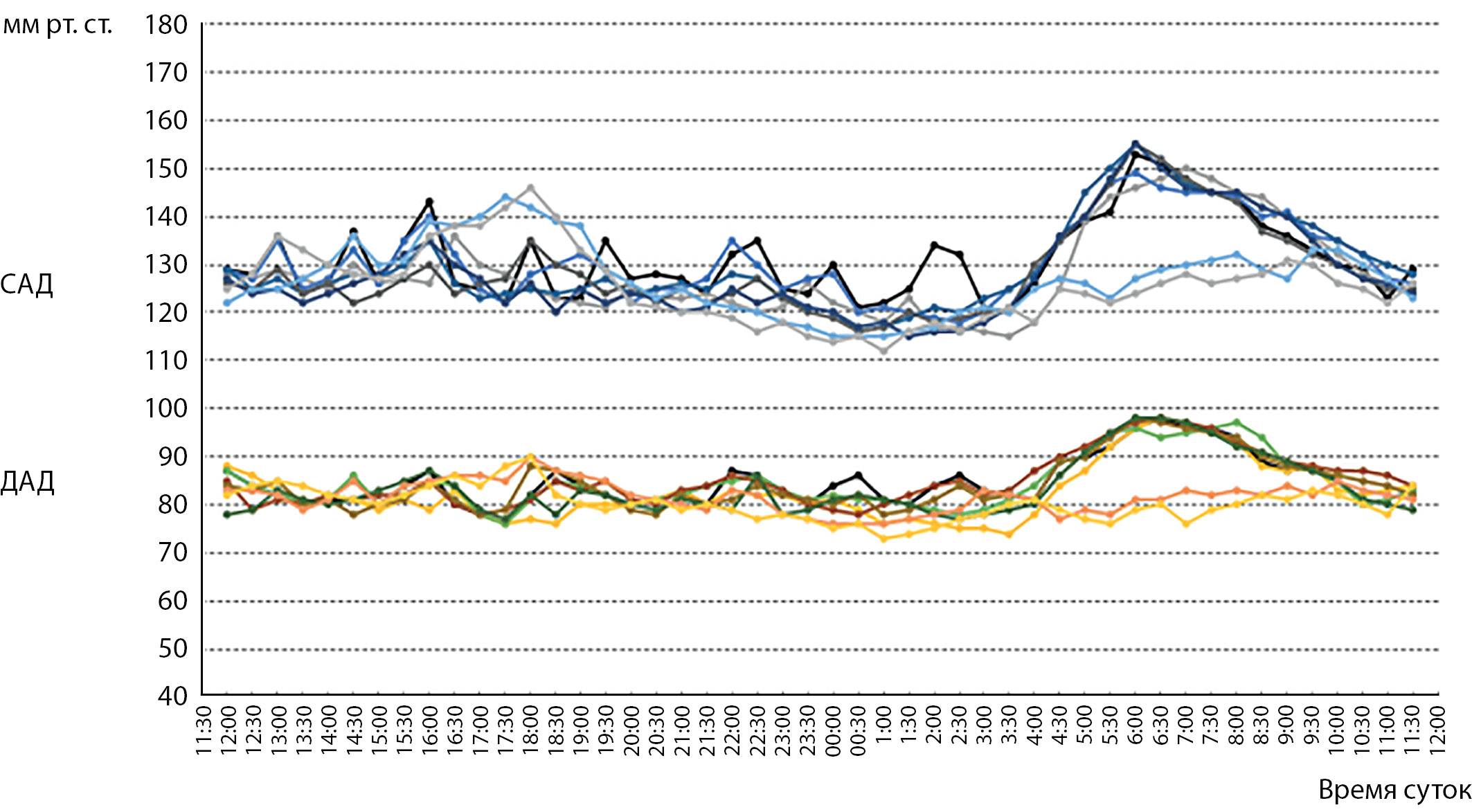
AIM. To reveal the peculiarities of steroidogenesis and arterial hypertension in «physiological» hyperandrogenism in men.
MATERIALS AND METHODS. One-stage simultaneous study. The groups of men with hyperandrogenism caused by increased total testosterone (n=34) and those with hyperandrogenism caused by increased dihydrotestosterone (DHT) (n=66) were compared. In determining the type of hyperandrogenism and allocating patients to groups, DHT and total testosterone levels were determined by enhanced chemiluminescence. Subgroups of men with and without arterial hypertension were compared in the group of patients with hyperandrogenism due to an increase in total testosterone. Body mass index, waist circumference, systolic and diastolic blood pressure, pulse, and LH, SBHG, estradiol, blood multisteroid levels by isotope dilution liquid chromatography/tandem mass spectrometry, glucose, blood lipid spectrum, uric acid, creatinine, renin, potassium, sodium, and blood chloride were assessed in all patients. Patients with arterial hypertension additionally underwent daily BP monitoring, albuminuria assessment, electrocardiography, ocular fundus examination. The baseline threshold level of significance was p<0.05. For multiple comparisons, the p significance level was calculated using the Bonferroni correction.
RESULTS. Statistically significant differences were found in the levels of 17-hydroxypregnenolone, 17-hydroxyprogesterone, and androstenedione, which were higher in men with elevated levels of total testosterone. No statistically significant differences in other laboratory parameters were found. No cases of increased blood pressure were detected in the group of men with elevated DHT. In the group of men with elevated total testosterone, 23,5% of men with arterial hypertension without targetorgan lesions were identified, while hyperandrogenism was associated with 17,6% of cases. Arterial hypertension associated with hyperandrogenism was characterized by a rise in blood pressure in the early morning hours. Estradiol levels, while remaining within normal limits, were statistically significantly lower in patients with arterial hypertension compared with men with elevated testosterone but without hypertension.
CONCLUSION. No cases of arterial hypertension were observed in «physiological» hyperandrogenism due to elevated DHT levels, whereas its incidence in «physiological» hyperandrogenism due to elevated total testosterone was 23,5%. The features of steroidogenesis were increased production of 17-hydroxypregnenolone, 17-hydroxyprogesterone, and androstenedione in men with testosterone hyperandrogenism and decreased estradiol production in patients with arterial hypertension compared with patients without testosterone hyperandrogenism.
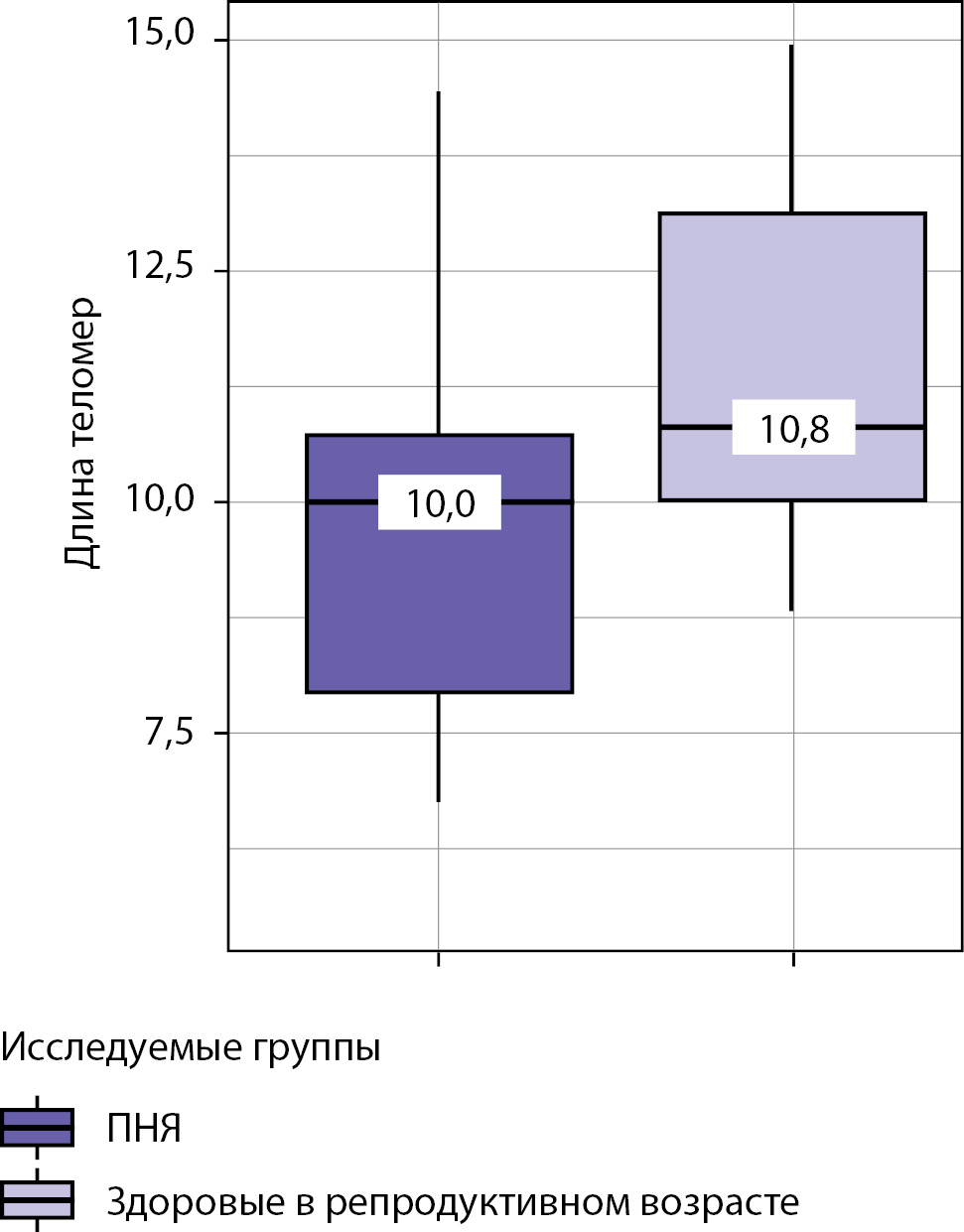
BACKGROUND: One of the most dangerous reproductive pathologies is primary ovarian insufficiency (POI). Except manifestation in the age <40 years old it leads to demographical losses, decrease of chances for healthy aging. POI can be characterized as summary of secondary amenorrhea, total estrogenic deficiency and hypergonadotropic hypogonadism. Hence, POI has probably harmful effect on telomere length. Telomere length determining and sex steroid replacement therapy may be promising and effective to prevent decrease of life quality/ longevity among females with POI.
AIM: To evaluate features of replicative (telomere length) and biochemical (metabolic syndrome) markers among females with primary ovarian insufficiency.
MATERIALS AND METHODS. Research has been provided in collaboration between Endocrinology Research Centre of the Russian Ministry of Health and Lomonosov Moscow State University Medical Research and Educational Centre in the period since 10.01.2021 until 01.08.2022. Females with non-iatrogenic hypergonadotropic hypogonadism caused by primary ovarian insufficiency (n=33); healthy females of reproductive age (18–49 y.o.; n=24). Patients have undergone laboratory genetic (leucocyte telomere length), biochemical analyses. DNA extraction — with Qiagen DNA blood mini kit (Germany).
Leukocyte telomere length — with real-time polymerase chain reaction PCR (Flow-fish). Soft program IBM SPSS Statistics (version 26,0 for Windows).
RESULTS. Females with POI due to estrogenic deficiency have slightly shorter mean telomere length (10,0 [7,9–10,7] kB, than healthy females of reproductive age (10,8 [10,0–13,1] кБ, р<0,001). Females with POI due to estrogenic deficiency have higher chances for development of carbohydrate metabolism disturbances (prediabetes) (р<0,043), increasement of FSH level (р<0,001). FSH level correlates moderately and negatively (ρ=0,434) with leukocyte telomere length (р<0,001).
CONCLUSIONS. Female with POI and receiving sex steroid replacement therapy have decrease of telomere length and increase of chances for carbohydrate metabolism disturbances in opposite to healthy reproductive females.

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND 4.0).
ISSN 2308-1430 (Online)






































