


Editorial
The recombinant technologies era, which began in the second half of the XX century, made it possible to produce recombinant growth hormone (rGH) necessary for the treatment of stunting of various genesis. The time of practically unlimited possibilities of rGH production has come, which served as a stimulus for studying the efficacy and safety of rGH application, searching for optimal ways of its use and dosing regimes. Many years of experience in the use of somatropin in clinical practice allowed us to obtain data on its effectiveness primarily in somatotropic insufficiency in children, to study its effect on the functional state of various organs and systems, and to expand the indications for the use of RGR.
Clinical endocrinology
Within the framework of the article, the authors analyzed the available information about the damage to the lacrimal apparatus during radionuclide therapy. In focus of article lesions of the lacrimal production system, the main and accessory lacrimal glands, as well as lacrimal drainage are considered. It was found that damage to the lacrimal apparatus is characteristic of 131I therapy for thyroid cancer, as well as for radioligand therapy using anti-PSMA antibodies labeled with 177Lu and 225Ac. 177Lu-PSMA and 225Ac-PSMA may damage the lacrimal gland with the formation of a clinically pronounced "dry eye syndrome". The pathogenesis of such lesions is associated with the accumulation of a radioisotope in the tissues of the lacrimal apparatus, while during therapy with 131I, accumulation is realized due to the expression of the sodium-iodine symporter in the nasolacrimal duct, and during therapy with 177Lu-PSMA and 225Ac-PSMA, the radiobiological effect is realized in connection with the expression PSMA by lacrimal tissue. An analysis of the available sources showed that to date there are no results of systematic studies on the problem, there is a lack of knowledge regarding the individual risks of developing these complications, methods for their prevention that have proven effectiveness have not been developed, and the treatment methods used, having relatively low efficiency, are not specialized. The authors concluded that the strengthening of interdisciplinary interaction, as well as the organization verification methodology and correct studies, can contribute to solving problems related to the study of the complications under consideration.
AIM: To develop a noninvasive method of differential diagnosis of ACTH-dependent hypercortisolism, as well as to evaluate the effectiveness of an optimal algorithm for predicting the probability of ectopic ACTH syndrome (EAS) obtained using machine learning methods based on the analysis of clinical data.
MATERIALS AND METHODS: As part of a single-center, one-stage, cohort study, a retrospective prediction of the probability of EAS among patients with ACTH-dependent hypercortisolism was carried out. Patients were randomly stratified into 2 samples: training (80%) and test (20%). Eleven machine learning algorithms were used to develop predictive models: Linear Discriminant Analysis, Logistic Regression, elastic network (GLMNET), Support Vector machine (SVM Radial), k-nearest neighbors (kNN), Naive Bayes, binary decision tree (CART), C5.0 decision tree algorithms, Bagged CART, Random Forest, Gradient Boosting (Stochastic Gradient Boosting, GBM).
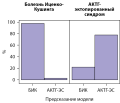
RESULTS: The study included 223 patients (163 women, 60 men) with ACTH-dependent hypercortisolism, of which 175 patients with Cushing’s disease (CD), 48 — with EAS. As a result of preliminary data processing and selection of the most informative signs, the final variables for the classification and prediction of EAS were selected: ACTH level at 08:00 hours, potassium level (the minimum value of potassium in the active stage of the disease), 24-h urinary free cortisol, late-night serum cortisol, late-night salivary cortisol, the largest size of pituitary adenoma according to MRI of the brain. The best predictive ability in a training sample of all trained machine learning models for all three final metrics (ROC-AUC (0.867), sensitivity (90%), specificity (56.4%)) demonstrated a model of gradient boosting (Generalized Boosted Modeling, GBM). In the test sample, the AUC, sensitivity and specificity of the model in predicting EAS were 0.920; 77.8% and 97.1%, respectively.
CONCLUSION: The prognostic model based on machine learning methods makes it possible to differentiate patients with EAS and CD based on basic clinical results and can be used as a primary screening of patients with ACTH-dependent hypercortisolism.
Primary glucocorticoid resistance (OMIM 615962) is a rare endocrinologic condition caused by resistance of the human glucocorticoid receptor (hGR) to glucocorticoids (GR) and characterised by general or partial insensitivity of target organs to GK. Compensatory activation of hypothalamic-pituitary-andrenal axis results in development of a various pathological conditions caused by overstimulation of adrenal glands. Clinical spectrum may range from asymptomatic cases to severe cases of mineralocorticoid and/or androgen excess. At present time, primary generalized glucocorticoid resistance has been exclusively associated with defects in the NR3C1 gene. Here, we present a case report of an adolescent patient with clinical presentation of glucocorticoid resistance confirmed by detailed endocrinologic evaluation but no confirmed mutations in the NR3C1 gene.
BACKGROUND: Primary hyperparathyroidism (PHPT) is a endocrine disorder characterized by excessive secretion of parathyroid hormone (PTH) from parathyroid gland tumors. Parathyroidectomy (PTE) is the main treatment for PHPT, but it can lead to hypocalcemia in up to 46% of cases. Hypocalcemia is associated with seizures and life-threatening cardiac arrhythmias, and vitamin D deficiency can exacerbate PHPT severity and contribute to «hungry bones syndrome,» resulting in severe and persistent postoperative hypocalcemia.
AIM: To evaluate the association and determine the strength of the relationship between preoperative cholecalciferol therapy and the occurrence of hypocalcemia within 1–3 days after PTE in patients with PHPT.
MATERIALS AND METHODS: The study was conducted at the Endocrinology Research Centre, during the periods of 1993–2010 and 2017–2020. The inclusion criteria consisted of patients diagnosed with PHPT who required PTE, had a serum 25-hydroxyvitamin D (25(OH)D) level below 20 ng/mL, and a serum total calcium level below 3 mmol/L. The exclusion criterion was the use of medications that affect calcium-phosphorus metabolism, including cinacalcet, denosumab, or bisphosphonates, either as monotherapy or as part of combination therapy.
RESULTS: There were 117 patients, including 110 (94%) females and 7 (6%) males. The median age and interquartile range were 58 [49; 65] years. Among the participants, 21 (18%) received cholecalciferol supplementation for a duration of 2 weeks to 2 months prior to PTE, aiming to address vitamin D deficiency. The remaining 96 (82%) participants did not receive cholecalciferol supplementation. Both groups, i.e., participants receiving cholecalciferol and those who did not, were similar in terms of anthropometric factors (sex and age at the time of surgery), preoperative clinical characteristics (BMD decrease), and laboratory parameters (PTH, total calcium, phosphorus, ALP, OC, CTX-1, and 25(OH)D levels). The occurrence of postoperative hypocalcemia was significantly lower in participants who received cholecalciferol supplementation (10% vs. 63%, p<0,001, FET2). Cholecalciferol intake showed a negative association with hypocalcemia development (RR=0,15, 95% CI (0,03; 0,51)).
CONCLUSION: Preoperative cholecalciferol supplementation for 2 weeks to 2 months before PTE reduces the risk of postoperative hypocalcemia in patients with PHPT by 2–33 times.
Relevance: Insulinoma is the most common hormonally active neuroendocrine tumor (NET) of the pancreas. In recent years, there has been a trend towards an increase in the incidence of NET especially insulinoma.
Aim: Summarizing and analyzing current data on various approaches to the treatment of insulinoma. Our review includes a comprehensive assessment of the advantages and disadvantages of currently available insulinoma treatment methods in comparison with past experience, as well as a review of promising methods that are not currently widely used.
Materials and methods: Analysis of literature from such databases as scientific electronic library elibrary.ru, Pubmed, Google Scholar, MedLine, Scopus and Web of Science.
Results: The most common treatment for insulinoma is surgery. For patients with high operative risk, alternative methods such as alcohol ablation, radiofrequency ablation, and tumor embolization may be used. Medications include the use of somatostatin analogues, diazoxide. The literature describes the potential benefit of the use of beta-blockers, phenytoin, glucagon, however, in clinical trials, these drugs have not demonstrated a significant effect. For the treatment of malignant and metastatically advanced insulinoma, targeted therapy (primarily Everolimus), chemotherapy, as well as embolization (including chemoembolization, radioembolization), radiofrequency ablation (RFA), microwave ablation and cryoablation, ultrasound ablation (HIFU), laser ablation, brachytherapy, irreversible electroporation are used.
Conclusion: The study of new drugs is an important task for scientists, among medications the most promising are new generations of somatostatin analogues, targeted drugs and chemotherapy drugs. The rare frequency of insulinoma makes it difficult to conduct randomized controlled trials and prospective studies. That is why physicians and scientists need to maintain close contacts with each other and take into account the experience of treating each patient with such disease, which will help develop effective treatment algorithms in the future.
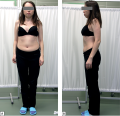
Hyperparathyroidism is a syndrome characterized by an excessive secretion of parathyroid hormone. Etiologically, hyperparathyroidism is subdivided into primary hyperparathyroidism, which develops as a result of parathyroid adenoma, carcinoma or hyperplasia, and secondary hyperparathyroidism, which happens as a compensatory response to a hypocalcemia caused by condition outside the parathyroid glands. Turner syndrome may also be accompanied by mineral metabolism disorders of various etiology. An association of hyperparathyroidism and Turner syndrome is interesting because of multifactorial impact on bone mineral density, but only few cases of such coexistence have been previously described in the literature. This article describes two patients with Turner syndrome and hyperparathyroidism of different etiology. Hyperparathyroidism, normocalcemia, vitamin D deficiency, osteoporosis, parathyroid tumors were found in both cases. In one case a number of assays was performed to confirm the patient’s normocalcemic primary hyperparathyroidism, and surgery was performed to achieve remission. In the second case, treatment of vitamin D deficiency resulted in normalization of serum concentration of parathormone, after which the patient was prescribed antiresorptive therapy. The pathogenetic association between Turner syndrome and hyperparathyroidism requires further investigation. Comprehensive approach to the diagnosis and treatment of mineral metabolism disorders are essential for patients with coexistence of these two diseases.
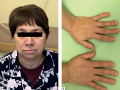
Acromegaly is a neuroendocrine disorder caused by excessive production of growth hormone (GH). In the majority of cases the cause of acromegaly is a pituitary tumor producing GH. Cases of ectopic acromegaly are much rarer. Ectopic acromegaly occurs in cases of tumors which produce growth hormone-releasing hormone (GHRH) or extrapituitary tumors which produce GH. The main sources of excessive GHRH production are neuroendocrine tumors (NETs) of the lung or pancreas. Treatment of ectopic acromegaly consists of surgical removal of the source of GHRH hyperproduction and in cases where surgery is not an option, somatostatin analogues, pegvisomant, chemotherapy, immunotherapy or radiation therapy are used.
In this article three cases of ectopic acromegaly due to GHRH-producing lung NETs are presented, each of them being notable for a number of features. In the first two cases, clinical symptoms were mild, besides in the second case ectopic acromegaly was accompanied by primary hyperparathyroidism. In the third case ectopic acromegaly was accompanied by pituitary macroadenoma, and after surgical removal of the lung NET remission of acromegaly was not achieved. In all three cases, lung NETs were detected incidentally on radiologic chest screening for other conditions.
Bones & Adipose tissues diseases

BACKGROUND: Multiple endocrine neoplasia type 1 (MEN1) — is a rare syndrome with an autosomal dominant inheritance pattern caused by a mutation in the tumor suppressor gene (MEN1). Parathyroid involvement is the most common MEN1 manifestation resulting in primary hyperparathyroidism (mPHPT). Data on the prevalence and structure of bone disease in mPHPT compared to sporadic one (sPHPT) are often incomplete and contradictory.
AIM: The purpose of this study was to compare the severity of bone involvement between mPHPT and sPHPT.
MATERIALS AND METHODS: A single-center retrospective study was conducted among young patients in the active phase of PHPT and without prior parathyroidectomy in anamnesis. The analysis included the main parameters of calcium-phosphorus metabolism, bone remodeling markers, as well as an assessment of disease complications. Bone mineral density (BMD) was measured using dual-energy X-ray absorptiometry (DXA) at sites of lumbar spine, femur and radius. Trabecular bone score (TBS) was applied to estimate trabecular microarchitecture. All patients included in the study underwent genetic testing.
RESULTS: Group 1 (mPHPT) included 26 patients, and group 2 (sSHPT) included 30 age-matched patients: the median age in group 1 was 34.5 years [25; 39], in group 2 — 30.5 years [28; 36], (p=0.439, U-test). Within group 1, the subgroup 1A (n=21) was formed with patients without other hormone-produced neuroendocrine neoplasms (NEN) in the gastrointestinal tract (GI) and the anterior pituitary gland. The duration of PHPT was comparable in both groups: mPHPT — 1 year [0; 3] versus sPHPT — 1 year [0; 1], (p=0.533, U-test). There were no differences in the main parameters of calcium-phosphorus metabolism, as well as in the prevalence of kidney complications. In the mPHPT group, bone abnormalities were observed significantly more often compared to sPHPT: 54 vs 10% (p=<0.001; F-test). Statistically significant differences were revealed both in BMD and in Z-score values of the femoral neck and total hip, which were lower in the mPHPT group. These differences remained significant when comparing subgroup 1A with sPHPT.
CONCLUSION: MEN1-associated PHPT may be accompanied by a more severe decrease in BMD in the femoral neck and total hip compared to sPHPT regardless of the other hormone-producing NEN. Clarifying the role of mutation in the MEN1 gene in these processes requires further study.
Carbohidrates metabolism disturbancies
The progressive nature of type 2 diabetes mellitus leads to the need for insulin therapy in a significant proportion of patients. Very often start of insulin therapy in type 2 diabetes mellitus (T2DM) is associated with weight gain and a significant increase of hypoglycemia’s risk. However, innovative options, such as fixed ratio combinations of glucagon-like peptide 1 receptor agonists (GLP-1RA) and basal insulin, minimize weight gain and hypoglycemia risks and allow a greater proportion of patients to achieve individual glycemic control goals without compromising safety parameters. This review includes a description of the randomized clinical trials, as well as the results of real clinical practice of the use of two currently existing fixed ration combinations of GLP-1RA and basal insulin — iDegLira and iGlarLixi.
Pediatric Endocrinology
The description of the child aged 5 months with the von Hippel-Lindau syndrome without any manifestations of this syndrome is presented. The reason for the molecular genetic examination was the presence of cases of this syndrome in the family (mother and sister). The heterozygous variant c.355T>C p.F119L was found in the VHL gene. An objective examination revealed no pathology. A comprehensive laboratory and instrumental examination aimed at searching for components of the von Hippel-Lindau syndrome, including a blood test for metanephrines and normetanephrines, ultrasound of the abdominal organs, examination of the fundus, also did not reveal any abnormalities. Given the results of molecular genetic diagnosis, the child remains under observation and will undergo regular examinations to identify components of the von Hippel-Lindau syndrome, including blood/urine tests for normetanephrines.
Clinical guidelines
On September 29, 2023, a meeting of the interdisciplinary expert council “Cognitive health of a comorbid patient” was held in Vladikavkaz. To reduce the social and economic burden of cognitive impairment, which is increasingly being detected in comorbid patients in the Russian Federation, it is necessary to introduce socially significant initiatives for the timely diagnosis and prevention of these diseases, as well as update modern approaches to treatment, taking into account their multifactorial pathogenesis and the risk of complications. Based on the results of scientific reports and discussions held during the expert council, experts made decisions on a further plan within the framework of socially significant initiatives for the prevention of obesity.

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND 4.0).
ISSN 2308-1430 (Online)





































