


Editorial
The Endocrinology Reserch Centre is proud not only of its achievements in the area of personalized approach to the examination and treatment of patients in accordance with the principles of evidence-based medicine, the use of modern diagnostic and treatment technologies, but also of its rich history of training scientific and medical personnel. For many years the Center has been attracting the best and most talented graduates of higher medical institutions, becoming the “alma mater” for young doctors - endocrinologists, pediatric endocrinologists and nutritionists. Specialists, graduates of NMRC, make a significant contribution to the development of medicine, conducting research, creating innovative methods of treatment and helping patients not only in our country, but also abroad. However, the State Research Center of the Russian Federation FGBU «NMRC Endocrinology» is famous not only for its high-quality and comprehensive training, but also for its experienced, dedicated teaching staff, giving students unique opportunities for professional growth. The purpose of this article is to reflect the most important milestones in the development of education at NMIC Endocrinology, which began its formation more than a century ago, during the most difficult period of transition for our country.
Clinical endocrinology
BACKGROUND: Amiodarone takes a leading position in arrhythmological practice in the prevention and relief of various cardiac arrhythmias. Type 2 amiodarone-induced thyrotoxicosis is a frequent side effect of the drug. It is the most complex type of thyroid dysfunction both in terms of the severity of clinical manifestations, and in terms of understanding the mechanisms of pathogenesis, possibility of differential diagnosis and providing effective treatment. Due to the increasing life expectancy of the population, corresponding increase in the frequency of cardiac arrhythmias, the problem does not lose its relevance. Identification of predictors, assessment and prediction of the individual risk of developing this thyroid pathology is a necessity in daily clinical practice for making a reasonable decision when prescribing the drug, determining the algorithm for further dynamic monitoring of the patient.
AIM: To evaluate the structure of amiodarone-induced thyroid dysfunction, prevalence, time and predictors of development type 2 amiodarone-induced thyrotoxicosis in a prospective cohort study.
MATERIALS AND METHODS: The study involved 124 patients without thyroid dysfunction who received amiodarone therapy for the first time. Evaluation of the functional state of the thyroid gland was performed initially, after prescribing the drug for the first 3 months 1 time per month, in the future – every 3 months. The follow-up period averaged 12-24 months. The end of the observation occurred with the development of amiodaron-induced thyroid dysfunction or patient's refusal to further participate in the study. For the differential diagnosis of the type of amiodarone-induced thyrotoxicosis, the level of anti-TSH receptor antibodies and thyroid scintigraphy with technetium pertechnetate were determined. The type and frequency of thyroid dysfunction, time and predictors of development type 2 amiodarone-induced thyrotoxicosis were evaluated.
RESULTS: The structure of amiodarone-induced thyroid dysfunction was represented by hypothyroidism in 19,3% (n=24), type 1 thyrotoxicosis in 1,6% (n=2), type 2 thyrotoxicosis in 23,4% (n=29). The median time of its development was 92,0 [69,0;116,0] weeks; the average period of common survival – 150,2±12,6 weeks (95% CI: 125,5–175,0), median – 144±21,7 weeks (95% CI: 101,4–186,6). The main predictors of type 2 amiodarone-induced thyrotoxicosis were: age (OR=0,931; 95% CI: 0,895–0,968; p<0.001), BMI (OR=0,859; 95% CI: 0,762–0,967; p=0,012), time from the start of amiodarone therapy (OR=1,023; 95% CI: 1,008–1,038; p=0,003). Age ≤60 years was associated with increased risk of the dysfunction by 2.4 times (OR=2,352; 95% CI: 1,053–5,253; p=0,037), BMI≤26,6 kg/m2 – 2,3 times (OR=2,301; 95% CI: 1,025–5,165; p=0,043).
CONCLUSION: The results allow to personalized estimate the risk of type 2 amiodarone-induced thyrotoxicosis and determine the patient's management tactic.

In recent years, a large number of studies have been carried out to research molecular genetic abnormalities in ACTH-secreting pituitary tumors. This review presents a comprehensive analysis of exome studies results (germline and somatic mutations, chromosomal abnormalities in corticotropinomas which developed as part of hereditary syndromes MEN 1, 2, 4, DICER1, Carney complex etc., and isolated tumors, respectively) and transcriptome (specific genes expression profiles in hormonally active and inactive corticotropinomas, regulation of cell cycles and signal pathways). Modern technologies (next-generation sequencing — NGS) allow us to study the state of the microRNAome, DNA methylome and inactive chromatin sites, in particular using RNA sequencing. Thus, a wide range of fundamental studies is shown, the results of which allow us to identify and comprehend the key previously known and new pathogenesis mechanisms and biomarkers of corticotropinomas. The characteristics of the most promising molecular genetic factors that can be used in clinical practice for screening and earlier diagnosis of hereditary syndromes and isolated corticotropinomas, differential diagnosis of various forms of endogenous hypercorticism, sensitivity to existing and potential therapies and personalized outcome determination of Cushing`s disease.

The progressive improvement of the classification using modern analytical methods is an essential tool for the development of precise and personalized approaches to the treatment of pituitary adenomas. In recent years, endocrinologists have witnessed evolutionary changes that have occurred in the histopathological identification of pituitary neoplasms, revealing new possibilities for studying tumorigenesis and predicting biological behavior.
The paper considers the historical aspects of the gradual improvement of the classification of pituitary adenomas, as well as the new international 2022 WHO classification, according to which pituitary adenomas are included in the list of neuroendocrine tumors (PitNETs) to reflect the biological aggressiveness of some non-metastatic pituitary adenomas. The characteristics of pituitary adenoma are presented, as well as a list of histological subtypes of aggressive neuroendocrine tumors of the pituitary gland, marked by the main potentials for invasive growth, an increased risk of recurrence and a negative clinical prognosis.
The expediency of changing the definition of «pituitary adenoma» to «neuroendocrine tumor» is discussed. It is emphasized that the introduction of a unified clinical, laboratory and morphological protocol into national clinical practice will help provide comparable comparative studies on the prognosis of the disease and the effectiveness of secondary therapy and also contribute to adequate management of potentially aggressive PitNETs.
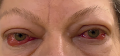
According to modern concepts, thyroid eye disease (TED) is an independent progressive autoimmune disease of the organ of vision, closely associated with the autoimmune pathology of the thyroid gland (TG), (ICD code — H06.2, proptosis in case of impaired thyroid function E05.0). TED treatment is a long step-by-step process, including immunosuppressive therapy, radiation therapy of the orbits and surgical treatment.
TED is a multidisciplinary problem. A patient with thyrotoxicosis clinic and TED symptoms will be taken to an endocrinological clinic for normalization of thyroid hormones and treatment of thyrotoxicosis complications. At the same time, under the supervision of an ophthalmologist, TED diagnostics and treatment will be carried out. Teamwork is of utmost importance because the effectiveness of TED treatment will depend on the speed of achieving a stable euthyroid state, the accuracy of determining the TED activity and severity, and the presence of complications requiring surgical treatment.
There are two main phases in the TED development. In the first phase of active inflammation, an increase in the symptoms of TED occurs, then a plateau phase follows, when the symptoms of activity persist but do not progress, then the symptoms regress and the process becomes inactive, while visual disturbances and cosmetic defects may persist. Determining the TED activity is very important from a clinical point of view, because the choice of treatment and tactics of patient management depend on the inflammation activity.
We describe a clinical case of phasing treatment of TED complicated by optic neuropathy and movement disorders in a patient with Graves’ disease, resistant to immunosuppressive therapy with glucocorticoids and requiring deep lateral bony orbital decompression.
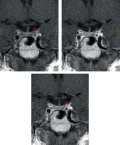
Endogenous hypercorticism (EH) is a severe symptom complex caused by hypercortisolemia; according to the etiology, ACTH-dependent and ACTH-independent variants are distinguished, which, according to the literature, occur in 70–80% and 20–30% of cases, respectively. A rare cause of ACTH-dependent endogenous hypercorticism is ACTH-ectopic syndrome (ACTH-ES) (about 15-20% of cases). ACTH-ES is a syndrome of adrenocorticotropic hormone (ACTH) hyperproduction by neuroendocrine tumors of extrahypophyseal origin. Various tumors can secrete ACTH: bronchopulmonary carcinoid, small cell lung cancer, less frequently, thymus carcinoid, islet cell tumors and pancreatic carcinoid, medullary thyroid cancer, carcinoid tumors of the intestine, ovaries, as well as pheochromocytoma (PCC).
This publication presents a clinical case of rarely detected paraneoplastic ACTH production by pheochromocytoma. The patient had clinical manifestations of hypercorticism, therefore, she applied to the Russian National Research Center of Endocrinology of the Ministry of Health of Russia. During the examination Cushing’s syndrome (CS) was confirmed, multispiral computed tomography (MSCT) of the abdominal cavity revealed a voluminous formation of the left adrenal gland. Additional examination recorded a multiple increase in urinary catecholamine levels. Subsequently, the patient underwent left-sided adrenalectomy. The diagnosis of pheochromocytoma was confirmed morphologically, immunohistochemical study demonstrated intensive expression of chromogranin A and ACTH by tumor cells.
Bones & Adipose tissues diseases
The main problem of obesity treatment is the difficulty of long-term weight maintenance. From one point of view, it can easily be explained by patients’ low compliance and absence of self-control. From another point of view, body weight is regulated not only by persons will, but also by multiple physiological mechanisms. Moreover, studies demonstrate that the attempts to reduce body weight stimulate the activation of adaptive biological process that block weight reduction.
Despite the variety of obesity treatment methods, only few patients are able to achieve significant (at least 5–7%) weight loss and maintain the result. In most cases people return to the initial weight in about 3–5 years. Therefore it is relevant to study weight regain mechanisms in order to identify new effective obesity treatment strategies.
The objective of this review is to summarize the information about the main issues of central, peripheral and behavioral pathogenic mechanisms which lead to disease relapse after obesity treatment and ideas for future strategies to resolve them.
Pediatric Endocrinology

BACKGROUND: Primary hyperparathyroidism (PHPT) is an endocrine disorder characterized by excessive secretion of parathyroid hormone (PTH) with upper-normal or elevated blood calcium levels due to primary thyroid gland pathology. PHPT is a rare pathology in children, with a prevalence of 2–5:100,000 children according to the literature. Due to the non-specificity of clinical manifestations at onset (nausea, vomiting, abdominal pain, emotional lability), the disease may remain undiagnosed for a long time.
AIM: To study the features of the course and molecular genetic basis of primary hyperparathyroidism in children.
MATERIALS AND METHODS: Retrospective observational study of 49 patients diagnosed with primary hyperparathyroidism. All patients underwent a comprehensive laboratory-instrumental and molecular genetic study at the Institute of Pediatric Endocrinology, Endocrinology Research Center of Russia in the period 2014–2022.
RESULTS: The first clinical symptoms of PHPT were noted at the age of 13.8 years [10.6; 1 5.2], among which fatigue, headaches, dyspepsia, lower limb pain, and fractures were the most common. The age of diagnosis was 15.81 years [13.1; 16.8], all children were found to have high levels of PTH, total and ionized calcium, with hypophosphatemia in 93.9% of patients (n=46) and hypercalciuria in 43% (n=21). Five out of 49 patients (10.2%) were found to have ectopy of the thyroid: 3 showed an intrathyroidal location, 2 in the mediastinal region. Molecular genetic study revealed mutations in 32.7% of patients (n=16, CI (21; 47)), mutations in MEN1 being the most frequent (n=11). Pathogenic variants in CDC73 were detected in 3 patients, RET — in 2. Among the operated 39 patients, adenoma of the thyroid was detected in 84.6% of cases (n=33), hyperplasia in 7.7% (n=3), atypical adenoma in 5.1% (n=2), carcinoma in 5.1% of cases (n=2).
CONCLUSION: The paper presents the peculiarities of the course and the results of molecular genetic study of pediatric PHPT. This sample is the largest among those published in the Russian Federation.
BACKGROUND: X-linked adrenoleukodystrophy (X-ALD) is a severe neurodegenerative metabolic disease with a frequency 1:17,000 in newborn boys. Being a major part of X-ALD with an incidence of 70–80% of patients, adrenal insufficiency (AI) is a life-threatening condition without timely treatment. The possibility of developing AI during the whole disease duration and the absence of any predictive factor for AI joining shows the necessity of studying AI in X-ALD patients to optimize current diagnostic and treatment algorithms.
AIM: To study diagnostic and therapeutic features of primary adrenal insufficiency due to X-ALD.
MATERIALS AND METHODS: A retrospective observational comparative study was conducted in 66 male patients, examined and treated in the Pediatric endocrinology department of Endocrinology Research Centre, Research Centre for Medical Genetics, Research and Clinical Institute for Pediatrics of the Pirogov Russian National Research Medical University Detached Structural Unit Russian Children’s Clinical Hospital (Moscow, Russia) for 2014–2022. All of patients were diagnosed with primary AI and a genetically confirmed X-ALD.
RESULTS: The median age of X-ALD manifestation was 6.6 years [4.7; 11.1]. The earliest age of AI diagnosis was 1.5 years at the preclinical stage and 1 year 8 months with clinical symptoms. The renin level was studied in 22.7% at the manifestation of AI (15/66 patients), mineralocorticoid deficiency was found in 7 patients. Family history was positive in 39.4% of patients (n=66), only in 15.1% (10/66 patients) of patients the disease was established at the preclinical stage. In 59.1% (n=66) the cerebral form of the disease (cALD) was established, in 16.6% — adrenomyeloneuropathy (AMN), and in 24.2% — isolated adrenal insufficiency (PAI). Age of AI establishment in the group of patients with AMN (15.6 years) significantly differs from the establishment of AI in patients with cALD (7.4 years, p=0.001) and PAI (5.6 years, p = 0.000). Mineralocorticoid therapy was prescribed simultaneously with glucocorticoid therapy in patients with cALD, in AMN and PAI patients it was added after 11 and 7 months, respectively (the differences between AMN and PAI groups were insignificant). Combined hormonal therapy receive 41% of patients with cALD, 54.5% of patients with AMN and 60% of patients with PAI.
CONCLUSION: It is necessary to examine all male patients with AI regardless of the manifestation age to exclude adrenoleukodystrophy, and it is also important to examine patients for the presence of AI regardless of X-ALD manifestation age. The assessment of renin level in the manifestation of AI is also needed to prescribe mineralcorticoid therapy timely. Studying family history is the main method to detect X-ALD at the preclinical stage.
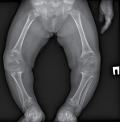
Growth retardation for more than 2 SD below the average population or presumed familial target height is classified as a short stature and may be a clinical manifestation of a large number of disorders. The use of the latest methods of molecular genetic analysis in recent years has allowed for a better understanding of the pathogenesis of inherited forms of a short stature. One of the recently discovered mechanisms of this pathology was monoallelic mutations in RPL13 gene, leading to the development of Isidor-Toutain type spondyloepimetaphyseal dysplasia (SEDM). Characteristic phenotypic features for this form are normal birth length, early postnatal growth deficiency, platyspondyly, proximal femoral epiphyseal changes, coxa vara, genu varum. This study presents the clinical and radiological characteristics of the first patient in the Russian Federation with SEMD caused by a mutation in RPL13 gene.
Reproductive Endocrinology
BACKGROUND: It has been suggested that the presence of chronic immunoinflammatory rheumatic disease (CIRD) may be a factor that increases the likelihood of developing hypogonadism syndrome, and conversely, the presence of uncompensated testosterone deficiency may predispose to a greater risk of developing or more severe course of ICRD.
AIM: To study the incidence of hypogonadism in men with rheumatoid arthritis (RA) and evaluate its impact on the course of RA and concomitant diseases.
MATERIALS AND METHODS: A one-time continuous study included 170 men with RA who were undergoing inpatient treatment at the Federal State Budgetary Institution NIIR named after. V.A. Nasonova. Patients were assessed for total testosterone levels and subsequently divided into subgroups with normal (>12 nmol/l) and reduced levels. An intergroup comparison was carried out on the main indicators used in clinical rheumatological practice to assess the stage, activity and other medical and demographic characteristics of RA, as well as the state of purine and carbohydrate metabolism. A correlation analysis was performed between the level of total testosterone and some clinical and laboratory parameters.
RESULTS: The frequency of detected testosterone deficiency in the study group was 24.1%. Significant correlations were noted between the level of total testosterone and body mass index (r=-0.29), the level of blood uric acid (r=-0.19) and C-reactive protein (r=-0.18). Patients with hypogonadism compared to the group with normal testosterone levels were characterized by higher body mass index (29.3±5.6 vs 26.3±4.0 kg/m2; p<0.001), glucose levels (6.95±7 .85 mmol/l vs 5.42±1.13 mmol/l; p=0.034) and uric acid (354.6±110.7 vs 317.5±84.8 µmol/l; p=0.03) blood. In addition, patients with hypogonadism were more likely to suffer from obesity (41.6% vs 15.7%; p=0.001) and diabetes mellitus (21.6% vs 10.2%; p=0.075) without a statistically significant difference, and also had higher ESR (46.5±42.2 vs 31.0±30.9 mm/h; p=0.012). A more frequent occurrence of anemia was noted in hypogonadism (32.4% vs 16.7%; p=0.041).
CONCLUSION: Testosterone levels and the presence of hypogonadism were not associated with the stage and activity of RA, however, testosterone deficiency was accompanied by a more frequent development of overweight and obesity, and a deterioration in purine and carbohydrate metabolism.

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND 4.0).
ISSN 2308-1430 (Online)





































